Blog
Orbion Team
Can You Predict Which Proteins Will Fight You at the Bench?

You have twelve targets and budget to seriously pursue four. Three months from now, some of these constructs will express cleanly, purify in a day, and behave. Others will sit in inclusion bodies, smear across your SEC column, or refuse to crystallize after 2,000 conditions. The painful part: you usually find out which is which only after you've spent the reagents.
Across structural genomics consortia, only a minority of cloned targets ever reach a solved structure — and at one major center, more than 60% of the total cost of structure determination went to attempts that failed [1, 2]. That failure is not random. A large fraction of it is written into the amino-acid sequence, and you can read it before you order a single primer.
Key Takeaways
Difficulty is partly predictable from sequence: Disorder, aggregation propensity, hydrophobicity, transmembrane content, and low-complexity regions correlate with downstream expression, solubility, and crystallization failure [1, 3].
Feasibility scores stratify success sharply: Proteins in the top 20% of a feasibility score reach roughly 62% production success versus 28% for the bottom 20%; for crystallization, 57% versus 10% [1].
No single feature decides it: A useful tractability score is a composite — solubility, disorder, aggregation, and membrane topology pooled together, not any one number in isolation [4, 1].
Triage beats heroics: Ranking targets by predicted difficulty up front lets you spend bench resources on the ones likely to pay off, and budget extra engineering for the ones that won't behave.
A prediction is a probability, not a verdict: Solubility predictors top out around 58–62% accuracy on hard benchmarks [5]. Use scores to rank and triage, not to kill targets outright.

Why Some Proteins Fight You — and Why It's Legible From Sequence
Every protein you push through expression and purification runs a gauntlet: transcription, translation, folding, solubility, stability in your buffer, and — if you need a structure — crystallization or grid behavior. A target fails when it stalls at any one of these. The structural-genomics consortia, which deliberately tracked both successes and failures in shared databases, gave the field something rare: large, honest datasets of what makes it to the end and what doesn't [1, 2].
The pattern from those datasets is consistent. An SG target has to clear every stage in turn — cloning, expression, purification, crystallization — and only a small fraction of initial attempts survive all the way to a solved structure; the failures cluster around a recognizable set of sequence and predicted-structure features [1]. Stage-specific success is low enough that more than 60% of the total cost of structure determination is spent on attempts that fail [1].
The core tension: the determinants of difficulty are biophysical and largely sequence-encoded, yet most target-selection decisions still get made on biological interest alone. You pick the protein because the biology is compelling, then discover its hydrophobicity index and disorder content were quietly stacked against you. Reading those features first doesn't change the biology — it changes how much resource and engineering you commit, and in what order.

The Sequence Determinants of Difficulty
When a target turns out to be hard, it's usually one or more of these five culprits. Each is predictable from sequence with established tools.
1. Intrinsic Disorder
Disordered regions don't fold into a stable structure, and they sabotage you at multiple stages: they expose protease-sensitive backbone, they resist crystallization by adding conformational heterogeneity, and long disordered stretches often correlate with poor expression. In the structural-genomics feasibility analyses, predicted disorder showed a strong negative correlation with crystallization success [1].
Disorder is one of the more tractable things to predict. IUPred estimates per-residue disorder from the pairwise interaction energy a residue can form — the insight being that disordered sequences are composed of amino acids that simply can't make enough stabilizing contacts to fold [6]. Energy-based and, more recently, language-model-based predictors give you a per-residue disorder track you can map onto the sequence.
What disorder prediction tells you: where the floppy regions are, how long they run, and whether they sit at the termini (often trimmable) or interrupt a domain (harder).
What it cannot tell you: whether a given disordered region is functionally essential, or whether it becomes ordered upon binding a partner.
Diagnostic question: Are the disordered stretches at the termini, or do they bisect the folded core? Terminal disorder is a construct-boundary problem you can engineer around. Internal disorder is a deeper liability.
2. Aggregation Propensity
Sequences with exposed hydrophobic, β-aggregation-prone segments stick to themselves. That shows up as inclusion bodies during expression, as precipitate during concentration, and as soluble aggregates that wreck your SEC profile.
TANGO predicts β-sheet-mediated aggregation from physicochemical principles, treating the aggregate core as fully buried β-structure. On a benchmark of 179 peptides it reached an 87% success rate and correctly called pathogenic versus protective mutations in the Alzheimer β-peptide, lysozyme, and transthyretin [3]. The practical output is a per-residue aggregation track: short, high-scoring hotspots are your risk regions.
What aggregation prediction tells you: where the sticky segments are, and whether a candidate mutation raises or lowers risk.
What it cannot tell you: your actual yield in a specific host and buffer — aggregation in vivo also depends on chaperones, expression rate, and conditions.
Diagnostic question: Are the aggregation hotspots buried in the folded core (lower practical risk) or exposed on the predicted surface (higher risk)?
3. Hydrophobicity and Surface Chemistry
Crystallization in particular is sensitive to surface chemistry. The structural-genomics data point to a sweet spot: proteins with a GRAVY (grand average of hydropathy) index near 0.1 crystallize best, and both very hydrophobic and very hydrophilic proteins underperform [1]. Extreme isoelectric points — strongly acidic or strongly basic — also correlate with lower success [1].
These are trivial to compute from sequence, yet they're among the most predictive single features in crystallization-propensity models. XtalPred combines nine such biochemical and biophysical features against distributions from TargetDB and bins the protein into one of five crystallization classes from "optimal" to "very difficult" [4].
Diagnostic question: Is your GRAVY far from ~0.1, or your pI in an extreme range? If so, expect surface-driven crystallization trouble — and consider surface-entropy reduction or fusion strategies.
4. Transmembrane Content
Membrane proteins are the canonical hard case. In the feasibility analyses, the presence of transmembrane helices was associated with a production success rate around 21% — roughly a third of the rate for soluble targets [1]. The overexpression of milligram quantities remains the central bottleneck in membrane-protein structural biology, and labs routinely screen many homologs just to find one well-behaved clone [7].
Predicting transmembrane topology from sequence is mature: you can estimate the number, position, and orientation of TM helices, which both flags the target as high-difficulty and tells you what kind of difficulty (a single-pass anchor is a different problem than a seven-TM bundle).
Diagnostic question: How many predicted TM helices, and is the target polytopic? Each added TM segment compounds the expression and solubilization challenge.
5. Low-Complexity and Compositional Bias
Low-complexity regions — runs of a few residue types, homopolymeric stretches, compositionally biased segments — interfere with folding and crystallization and frequently overlap with disorder. XtalPred explicitly screens for low-complexity and coiled-coil regions as part of its difficulty assessment [4]. A long coiled-coil signals a non-globular shape that crystallizes on its own terms, if at all.
Diagnostic question: Does the sequence carry long low-complexity or coiled-coil stretches that aren't part of the domain you actually care about? If so, a tighter construct boundary may rescue tractability.
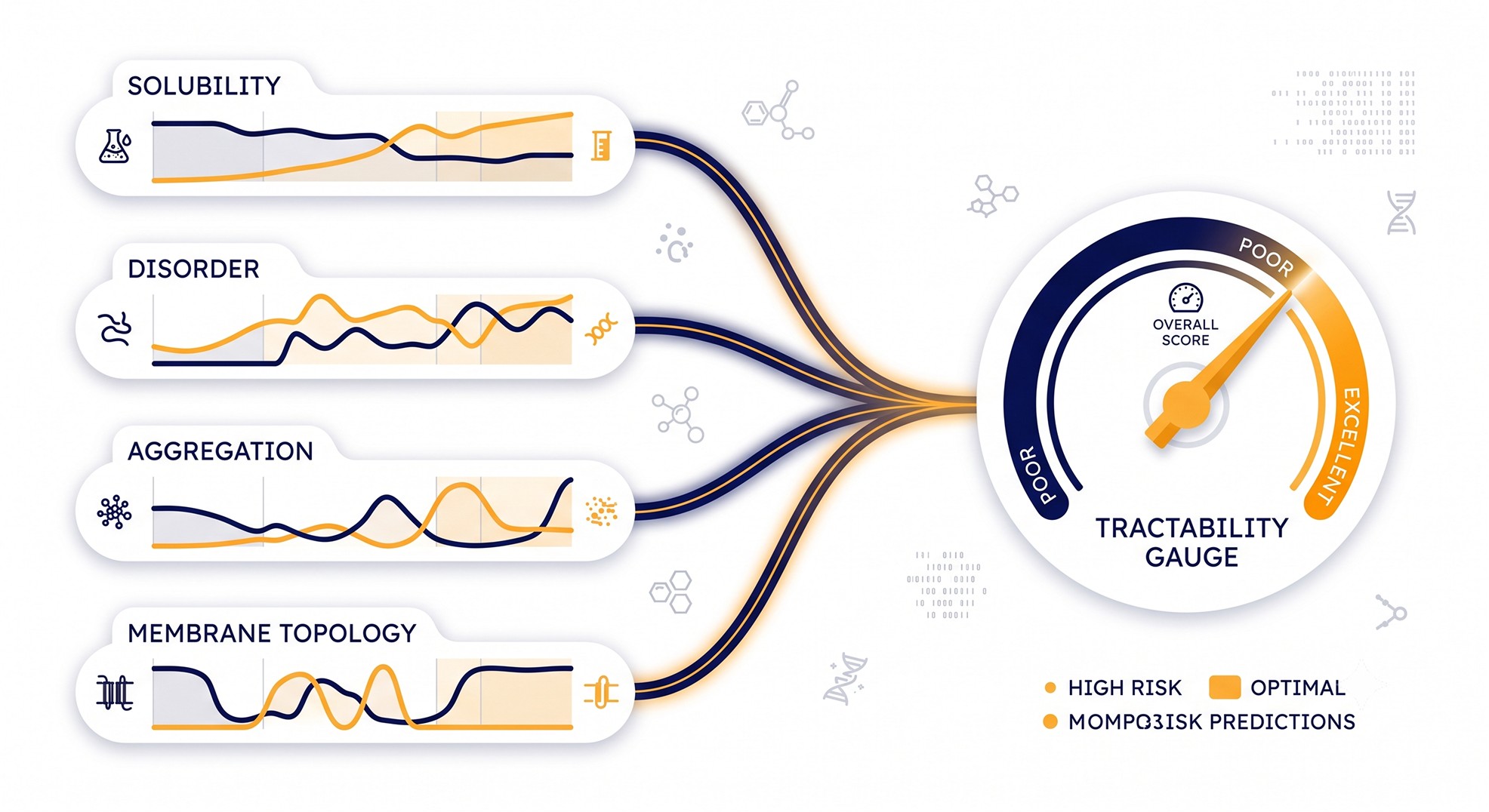
From Features to a Composite Tractability Score
No single feature is decisive. A protein can be perfectly ordered and still fail on a hydrophobic surface; another can carry a disordered tail and crystallize fine once you trim it. The structural-genomics community's durable lesson is that you get predictive power by pooling features, not by ranking on any one.
XtalPred's crystallization feasibility score does exactly this — it uses a logarithmic opinion pool to combine the probability distributions of individual features into one score, then assigns a difficulty class [4, 1]. The validation is the part worth internalizing: proteins scoring in the top 20% for production feasibility reached ~62% success versus ~28% for the bottom 20%; for crystallization, the top class hit ~57% versus ~10% for the worst [1]. That's a 2–6× spread in success probability, available before you touch a bench.
A practical composite for target tractability scoring draws on four predicted layers:
Layer | Predicts | Representative tools | Failure mode flagged |
|---|---|---|---|
Solubility / expressibility | Soluble yield in a host | SoluProt, CamSol, NetSolP [5, 8] | Inclusion bodies, no expression |
Disorder | Unfolded regions | IUPred and successors [6] | Proteolysis, heterogeneity |
Aggregation | Sticky β-prone segments | TANGO [3] | Precipitation, soluble aggregates |
Membrane topology | TM helices, orientation | Topology predictors [7] | Low yield, detergent dependence |
Pool these into one number and you have a defensible protein expression difficulty prediction you can sort a target list on. The point isn't a precise probability — it's a ranking that's far better than biological interest alone.
How Good Are These Predictions, Really?
Honest answer: good enough to triage, not good enough to be a verdict. Sequence-based solubility prediction is genuinely hard. SoluProt, trained on the TargetTrack database, reported 58.5% accuracy and an AUC of 0.62 on a balanced independent test set — ahead of PROSO II, SWI, and CamSol, but a long way from a coin flip becoming a certainty [5]. CamSol's intrinsic solubility profile correlates strongly with in vivo protein-abundance data, which is exactly what you want for ranking, but it still can't promise your specific construct will behave [8].
So calibrate your expectations:
Use scores to rank, not to kill. A target in the "very difficult" class isn't doomed; it's a signal to budget extra engineering — fusion tags, homolog screening, tighter boundaries — or to sequence it later in the queue.
Trust composites over single features. Any one predictor at ~60% accuracy is noisy. Agreement across disorder, aggregation, solubility, and topology is far more reliable than any single track.
Predictions don't model your host or buffer. In vivo solubility depends on chaperones, induction temperature, and codon usage that a sequence predictor never sees.

A Triage Decision Tree

Case Study: Triaging a Six-Target GPCR-Adjacent Panel
Problem: A discovery team had six candidate targets for a structure-enabled campaign and budget to fully resource two. Picking on biological priority alone, they'd have started with the two most therapeutically interesting — both polytopic membrane proteins.
Analysis: A sequence-first triage changed the order. Running disorder, aggregation, hydrophobicity/GRAVY, and topology predictions across all six produced a clear stratification. Two targets were polytopic membrane proteins (multiple predicted TM helices) — by the structural-genomics priors, production success in the ~20% range and a long road on solubilization [1, 7]. Two were soluble but carried long internal disordered regions and surface aggregation hotspots — fixable, but only with construct redesign. The remaining two were soluble, well-ordered, GRAVY near 0.1, moderate pI — squarely in the "likely tractable" class.
Solution: The team re-sequenced the queue. They started the two tractable targets immediately on a standard pipeline, put the two disordered targets through construct-boundary redesign and re-scored the trimmed constructs before committing, and routed the two membrane targets to a parallel homolog-screening and cryo-EM-oriented track rather than chasing crystals.
Outcome: The two tractable targets reached purified, monodisperse protein on the first construct — consistent with the ~57–62% success rates the top feasibility class predicts [1]. One redesigned (trimmed) construct moved from a smearing SEC profile to a single peak. The membrane targets stayed hard, as expected, but the team had budgeted for that difficulty up front instead of burning their first quarter on it. Net effect: bench effort concentrated where the sequence said it would pay off, and the hard targets got the right strategy from day one rather than after three failed months.
Practical Checklist
Before you commit bench resources to a target, verify:
Predicted transmembrane topology — how many TM helices, polytopic or single-pass?
Disorder track mapped onto the sequence — terminal (trimmable) or internal?
Aggregation hotspots identified — buried in core or exposed on surface?
GRAVY index and pI computed — near the favorable window (~0.1, moderate pI) or extreme?
Low-complexity / coiled-coil regions flagged outside your domain of interest
A composite tractability rank across all targets, not just per-target scores
Construct boundaries re-scored after any planned truncation
An explicit difficulty budget — which targets get extra engineering, which get queued late
The Economics of Triaging Up Front
Approach | Up-front time | Bench cost per dead target | Effect on attrition |
|---|---|---|---|
Biology-only selection | None | Full (months of cloning, expression, purification) | Baseline — most cloned targets fail before a structure [1] |
Single-feature filter (e.g., GRAVY only) | Minutes | Reduced, but misses multi-factor failures | Modest improvement |
Composite sequence triage | Hours | Avoided on low-ranked targets before reagents spent | 2–6× success spread between top and bottom feasibility classes [1] |
ROI consideration: with more than 60% of structure-determination cost going to failed attempts [1], even an imperfect ~60%-accurate triage that reorders your queue pays for itself the first time it stops you from sinking a quarter into a "very difficult" target you could have queued last. You're not trying to predict the future perfectly. You're trying to spend your best months on your best odds.
Bottom Line
Experimental difficulty is largely written into the sequence — disorder, aggregation, hydrophobicity, and transmembrane content predict who will fight you at the bench. Score your targets before you commit reagents, rank them by a composite tractability score, and spend your bench budget top-down. The predictions are noisy, so use them to triage and to budget engineering effort, never to kill a target outright.
How Orbion Helps
Reading these five determinants off a sequence by hand means stitching together half a dozen separate web servers and reconciling their outputs. Orbion folds them into the workflow you're already running on the target.
In the Characterization module, AstraUNFOLD generates the residue-level tracks that drive difficulty assessment directly on your sequence — transmembrane helix topology, per-residue disorder probability, and per-residue amyloidogenicity, all mapped onto the structure alongside pLDDT and PAE confidence. That's three of the four composite layers in one place, so you can see at a glance whether a disordered stretch sits at a trimmable terminus or bisects the core, and whether aggregation hotspots are buried or surface-exposed.
When you move to the Bench module to plan experiments, Orbion's Rate of Ease score puts a number on exactly the question this article is about: it's an Orbion score indicating predicted experimental difficulty for a given approach, derived from solubility, disorder, aggregation, and membrane topology predictions — the same composite logic the structural-genomics community validated, applied to your specific construct and goal.
Relevant Orbion features:
AstraUNFOLD (Characterization): per-residue disorder, amyloidogenicity, and transmembrane topology tracks — the raw determinants of difficulty, read straight from sequence.
Rate of Ease (Bench): a composite predicted-difficulty score over solubility, disorder, aggregation, and membrane topology, scoped to your chosen approach.
Design module construct scoring: when a target flags as hard, re-score trimmed or fusion-tagged constructs on solubility, disorder, and aggregation before you commit — so you triage and redesign in one loop instead of discovering the boundary problem at the bench.
Upload a sequence, read the difficulty before you order primers, and put your best months on your best odds.
References
Slabinski L, Jaroszewski L, Rodrigues APC, Rychlewski L, Wilson IA, Lesley SA, Godzik A. (2007). The challenge of protein structure determination—lessons from structural genomics. Protein Science, 16(11):2472-2482. Link
Price WN 2nd, Handelman SK, Everett JK, et al. (2009). Understanding the physical properties that control protein crystallization by analysis of large-scale experimental data. Nature Biotechnology, 27(1):51-57. Link
Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. (2004). Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nature Biotechnology, 22(10):1302-1306. Link
Slabinski L, Jaroszewski L, Rychlewski L, Wilson IA, Lesley SA, Godzik A. (2007). XtalPred: a web server for prediction of protein crystallizability. Bioinformatics, 23(24):3403-3405. Link
Hon J, Marusiak M, Martinek T, Kunka A, Zendulka J, Bednar D, Damborsky J. (2021). SoluProt: prediction of soluble protein expression in Escherichia coli. Bioinformatics, 37(1):23-28. Link
Dosztányi Z, Csizmók V, Tompa P, Simon I. (2005). IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics, 21(16):3433-3434. Link
He Y, Wang K, Yan N. (2014). The recombinant expression systems for structure determination of eukaryotic membrane proteins. Protein & Cell, 5(9):658-672. Link
Sormanni P, Aprile FA, Vendruscolo M. (2015). The CamSol method of rational design of protein mutants with enhanced solubility. Journal of Molecular Biology, 427(2):478-490. Link
Book a 20-Minute Demo
Sign up free for unlimited Overview runs — summary, sequence-based analysis, homology search. For the full Characterization — PTMs, binding sites, stability variants, construct design — book a demo and we'll run your target live.
