Blog
Orbion Team
Reading Membrane Protein Topology From Sequence Alone

You inherited a transporter project with a 12-helix prediction from a 2009 paper. You designed a C-terminal His-tag construct, expressed it, and got nothing on the Ni column — because the C-terminus you tagged sits in the periplasm, not the cytoplasm where your tag could fold and bind. A single wrong call on in/out orientation cost you a cloning round, an expression run, and three weeks.
Membrane proteins are roughly 27% of the human proteome and the majority of drug targets, yet most never yield an experimental structure. For these proteins, the only thing you reliably have is the sequence — and what you can read out of it is the topology: how many transmembrane (TM) helices, where they sit, and which loops face in versus out.
Key Takeaways
Topology is the cheapest high-value prediction you can make: number and position of TM helices plus in/out orientation, all from sequence, before you commit a single primer.
The positive-inside rule does the orienting: cytoplasmic loops carry more Arg and Lys than extracellular loops — this charge bias, not hydrophobicity, fixes which side faces in.
Methods evolved in three jumps: Kyte–Doolittle hydropathy (helix count only) → HMMs like TMHMM and Phobius (count + orientation, signal peptide handling) → deep learning like DeepTMHMM (single model, higher accuracy across α-helical and β-barrel proteins).
Topology drives construct design: tag placement, truncation boundaries, fusion choice, and signal-peptide strategy all hinge on which terminus and which loops are accessible.
Predictors still fail in three places: re-entrant loops (half-membrane dips), marginally hydrophobic helices, and dual-topology proteins that insert both ways — treat these calls as hypotheses, not facts.
Why Topology Matters Before You Touch a Pipette
A soluble protein has one job for construct design: get it to fold and stay soluble. A membrane protein adds a second axis — orientation in the bilayer — and that axis silently breaks experiments when you get it wrong.
Consider what topology actually tells you:
Which terminus is accessible from the cytoplasm. A cytoplasmic His-tag folds and binds Ni-NTA. A periplasmic or extracellular His-tag often doesn't fold properly in the oxidizing compartment and can be clipped by proteases.
Where the loops are. Want to insert a BRIL or T4 lysozyme fusion to aid crystallization? It goes in a cytoplasmic loop, not a tight periplasmic turn.
Whether you need a signal peptide at all. A protein whose N-terminus belongs outside needs translocation machinery engaged correctly; the wrong assumption gives you cytoplasmic inclusion bodies.
Where you can safely truncate. Cutting in the middle of a TM helix to make a soluble domain construct leaves a hydrophobic stub that aggregates.
The core tension: the experimental cost of a wrong topology call is high and the prediction is nearly free. Spend ten minutes reading topology from sequence and you avoid weeks of debugging a construct that was doomed at the design stage.

What "Topology" Means, Precisely
When people say membrane protein topology prediction, they bundle four distinct outputs. Keep them separate, because predictors differ in how well they handle each:
TM helix count and boundaries — how many membrane-spanning segments, and the residue ranges. This is the oldest, easiest target.
In/out orientation — for each loop and terminus, cytoplasmic ("in") or extracytoplasmic ("out"). This is where the positive-inside rule earns its keep.
Signal peptide vs. signal anchor — is that hydrophobic N-terminal stretch a cleavable signal peptide, or a real first TM helix (signal anchor) that stays in the membrane?
Irregular features — re-entrant loops, half-helices, kinks. The hardest, least reliable predictions.
A clean topology call answers all four. Most tools nail the first two for canonical α-helical proteins and degrade on the last two.

From Hydropathy to Deep Learning: How the Methods Evolved
Reason the field started: hydropathy plots (Kyte–Doolittle, 1982)
The original insight is simple and still useful. A TM helix is ~20 hydrophobic residues in a row. Slide a window across the sequence, average a hydrophobicity scale over each window, and plot it. Peaks above a threshold are candidate TM helices.
Kyte and Doolittle showed that a window of 19–21 residues makes TM domains "stand out sharply," with central values of at least 1.6 and peaks above 1.8 marking likely membrane spans [1].
What hydropathy can do:
Count candidate TM helices fast
Give you a visual, interpretable plot
Flag obviously hydrophobic stretches
What hydropathy cannot do:
Assign in/out orientation (it has no charge model)
Distinguish a signal peptide from a TM helix
Catch marginally hydrophobic helices that dip below threshold
Handle re-entrant loops at all
Hydropathy gives you helix count, not topology. For orientation you need a second principle.
The orienting principle: the positive-inside rule (von Heijne, 1992)
Gunnar von Heijne's observation reshaped the field: loops on the cytoplasmic side of the membrane are enriched in the positively charged residues arginine and lysine relative to loops on the outside. Charged residues resist translocation across the bilayer, and the membrane potential reinforces the bias.
Von Heijne turned this into an algorithm: generate candidate topologies from hydropathy, then rank them by how well the charge distribution matches positive-inside. On 24 bacterial inner-membrane proteins with experimentally known topology, this predicted the correct topology for 23 and identified 135 TM segments with only one overprediction [2]. This is the logic behind TOPPRED and every serious predictor since.
Diagnostic question: count Arg + Lys in your predicted cytoplasmic loops versus your predicted extracellular loops. If the "outside" loops are more positive, your orientation is probably flipped.
The HMM era: TMHMM (2001) and Phobius (2004)
Hidden Markov models unified counting and orientation into one probabilistic grammar. TMHMM models the protein as a sequence of states — helix core, helix caps, cytoplasmic loop, non-cytoplasmic loop — each with its own amino acid distribution, and finds the most probable labeling of your sequence.
TMHMM correctly predicts 97–98% of TM helices and discriminates membrane from soluble proteins with specificity and sensitivity above 99% [3]. Its one well-known weakness: accuracy drops when a signal peptide is present, because a cleavable signal peptide looks almost identical to a TM helix.
Phobius fixed exactly that. By modeling signal peptides and TM topology in one combined HMM, it resolves the cross-talk between the two [4]. Across whole proteomes, conventional separate predictors (SignalP + TMHMM) produced overlapping predictions affecting 25–35% of TM topologies and 30–65% of signal peptides — and since only one method can be right for each overlap, the combined model removes that ambiguity [10].
Why this matters for you: if your protein has a real N-terminal signal peptide and you run a TM-only predictor, you may count one phantom TM helix and flip the orientation of everything downstream. Use a tool that models both.
The deep-learning era: DeepTMHMM (2022)
The current generation replaces hand-built HMM states with protein language model embeddings feeding a neural network. DeepTMHMM, from the same DTU lineage as TMHMM, predicts topology for both α-helical and β-barrel membrane proteins in a single model, labels each residue, and handles signal peptides — reaching higher accuracy than the HMM tools across diverse proteomes [5].
DeepTMHMM emits a per-residue label from a small alphabet (inside, TM helix, outside, signal peptide, and β-strand / periplasm labels for barrels). That per-residue output is exactly what you want for construct design: you can read tag-safe loops and truncation points straight off the labeling.
The honest caveat: deep learning raised the floor but didn't eliminate the hard cases below. A higher headline accuracy still masks systematic blind spots.
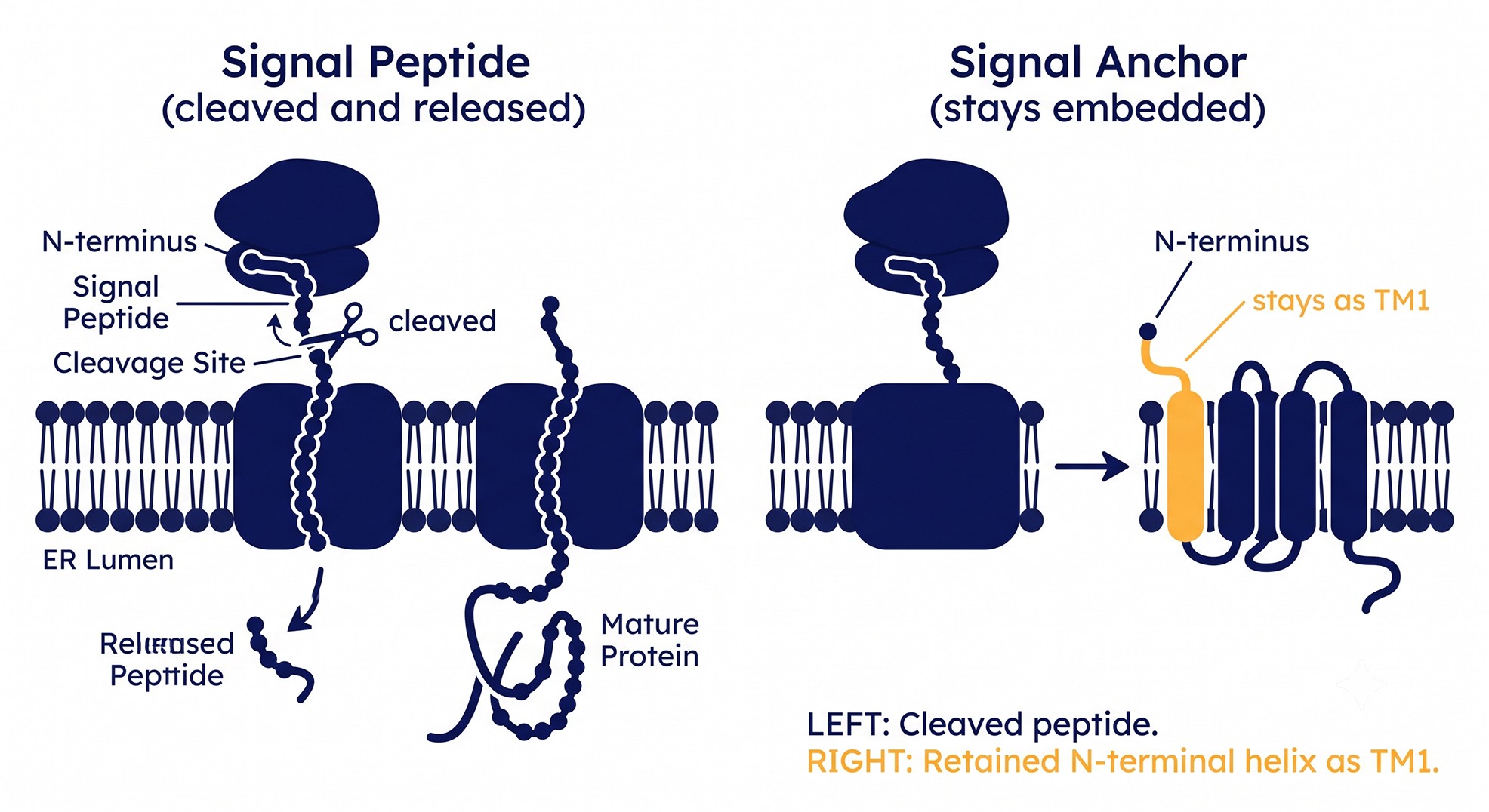
Signal Peptide vs. Signal Anchor: The Mistake That Flips Everything
This deserves its own section because it's the single most common topology error in construct design.
A signal peptide is a cleavable N-terminal hydrophobic stretch that targets the protein for translocation and is then cut off by signal peptidase. A signal anchor is an N-terminal hydrophobic helix that does the same targeting job but stays in the membrane as TM helix 1. They look nearly identical to a hydropathy plot — both are ~20 hydrophobic residues at the N-terminus.
Get it wrong and:
You count one too many (or too few) TM helices.
You flip the in/out orientation of every loop downstream of it.
You design a construct that either keeps a signal peptide you should have removed, or removes a TM helix you needed.
SignalP 6.0 — built on a protein language model — distinguishes all five signal-peptide classes and, run alongside a topology predictor, helps you separate "cleaved and gone" from "stays as TM1" [6]. When the topology of your whole protein seems to flip between tools, the N-terminal hydrophobic segment is usually the culprit. Resolve it first.
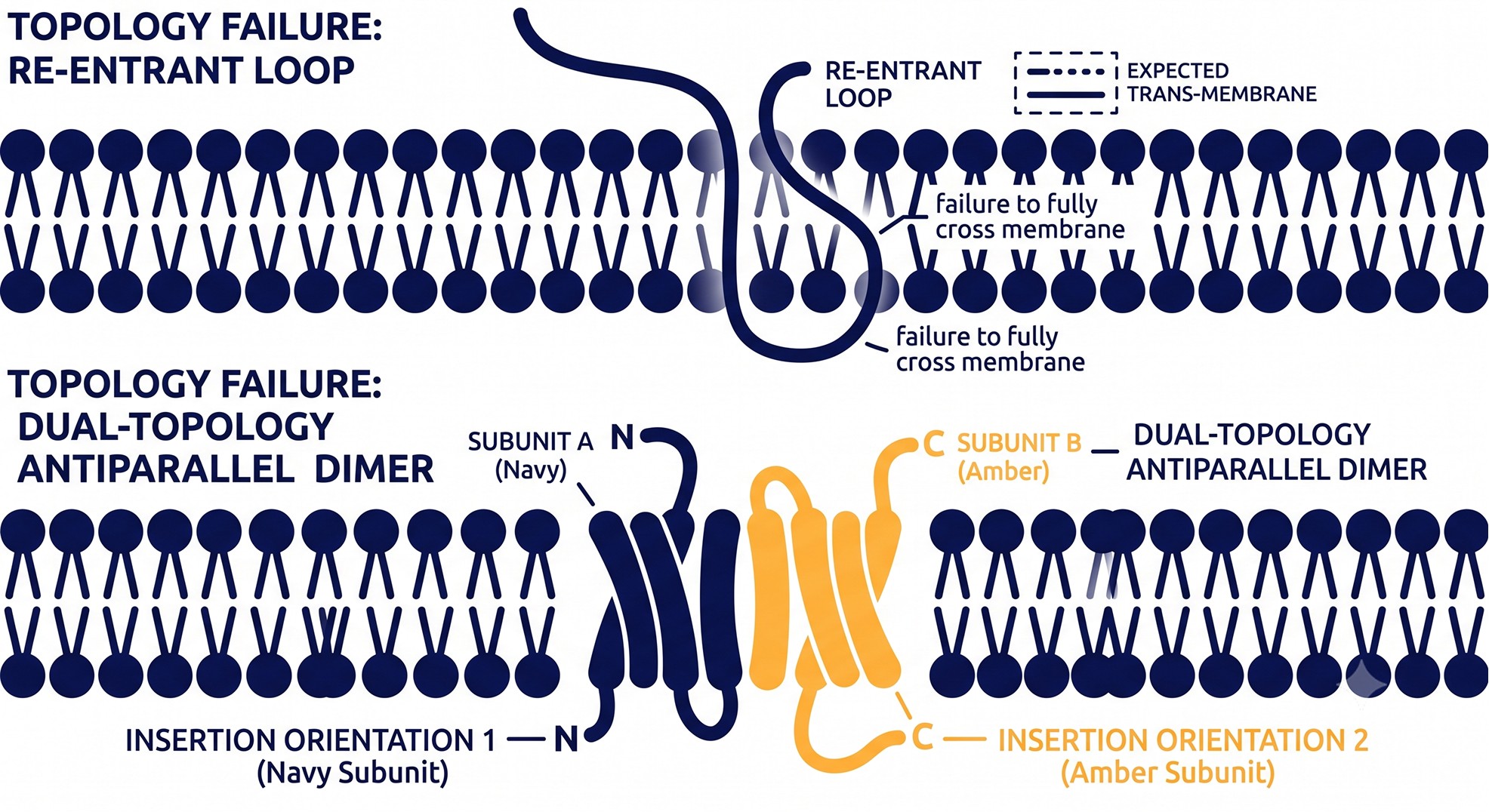
Where Predictors Still Fail
Treat every topology prediction as a confidence-weighted hypothesis. Three failure modes break even modern tools.
Re-entrant loops and half-helices
A re-entrant loop dips into the membrane from one side and comes back out the same side — it never crosses. Aquaporins are the textbook case: six full TM helices plus two half-helices that dip in and form the pore's selectivity filter. Ion channels (the P-loop) and many transporters have them too.
Hydropathy and standard HMMs are built to detect full membrane crossings, so a re-entrant loop is either missed entirely or miscounted as a full TM helix — which then flips downstream orientation. Re-entrant regions vary widely in length and depth but are surprisingly homogeneous in amino acid composition, which is why dedicated classifiers were needed to catch them at all [7]. If your protein is a channel, transporter, or aquaporin family member, assume re-entrant loops are present and verify against any available homolog structure.
Marginally hydrophobic helices
Not every functional TM helix is a clean hydrophobic block. Some are marginally hydrophobic — they sit below the usual threshold and only insert into the membrane with help from neighboring helices during folding. Hydropathy misses them; HMMs often do too. The result is an undercount, which again propagates an orientation error through the rest of the chain.
Dual topology
Some small membrane proteins insert into the bilayer in both orientations at once. The classic example is EmrE, a 110-residue, 4-TM small multidrug-resistance transporter that assembles as an antiparallel dimer — one subunit Nin/Cin, the other Nout/Cout [8]. The positive-inside rule is weak here by design: these proteins evolved a near-neutral charge balance so neither orientation dominates. A single-answer predictor will confidently report one orientation and be half wrong.
Diagnostic question: is your protein small (under ~120 residues), with few charged residues in its loops, in the SMR or a related family? If so, distrust any confident single-orientation call and check the literature for dual-topology evidence.
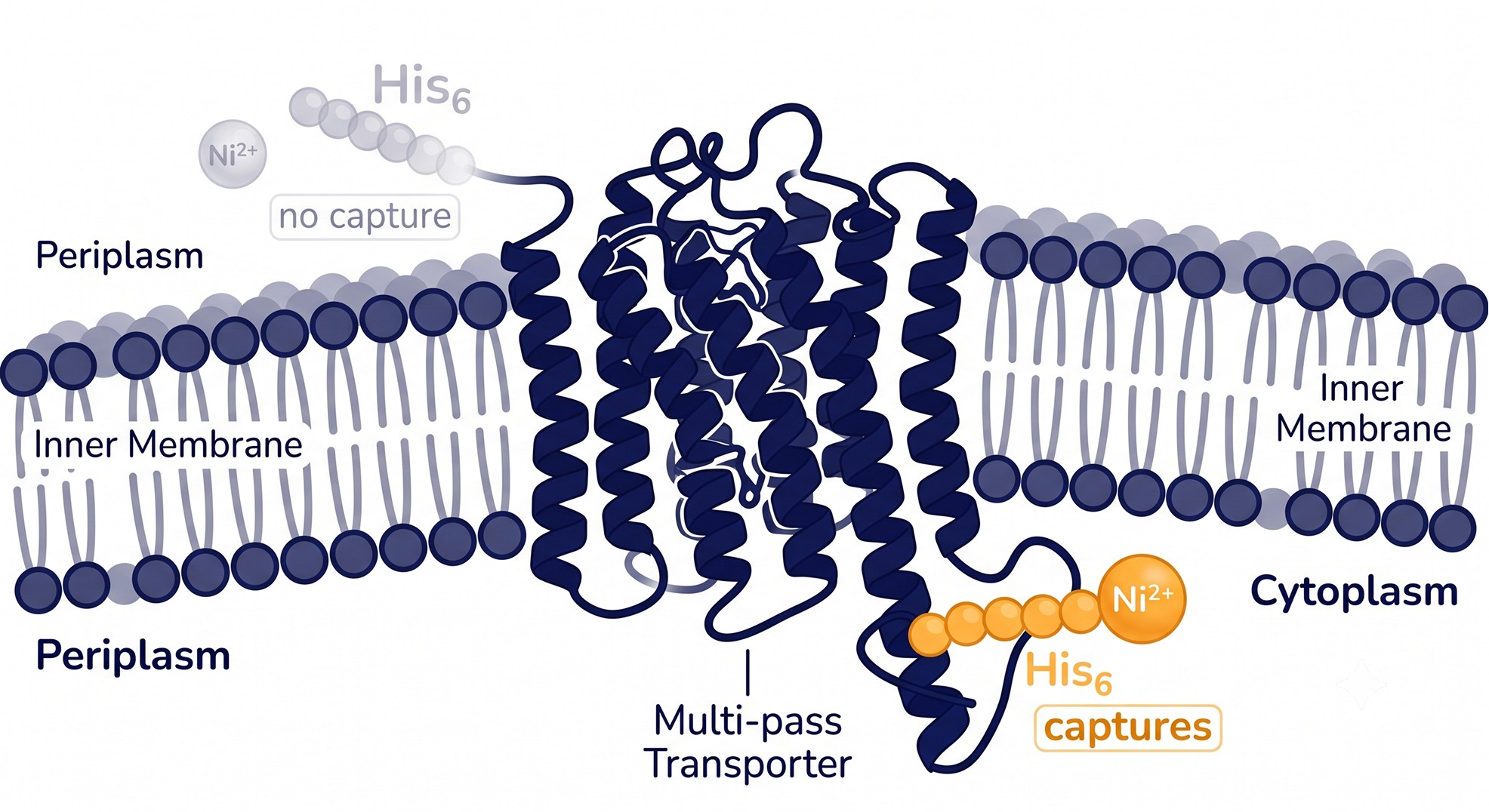
Case Study: Rescuing a Mis-Tagged Transporter Construct
Problem: A secondary active transporter (predicted 12 TM helices) expressed at the membrane but gave near-zero yield on Ni-NTA affinity purification. The team had used a C-terminal His10 tag because their soluble-protein default was C-terminal tagging.
Analysis: Re-running the sequence through a modern topology predictor returned 12 TM helices with both N- and C-termini facing the periplasm (Nout/Cout). The original 12-helix call had been correct on count but the team never checked orientation. A periplasmic His-tag in E. coli sits in the oxidizing periplasm, folds poorly, and is exposed to periplasmic proteases — explaining the failed capture. Checking the positive-inside balance confirmed it: the cytoplasmic loops carried a clear Arg/Lys excess consistent with the predicted in/out pattern.
Solution: Both termini were periplasmic, so neither could host a working E. coli His-tag. Using the per-residue labeling, the team identified a generously sized cytoplasmic loop and inserted a small internal affinity tag there, paired with a cytoplasmic BRIL fusion to aid downstream crystallization. They added a TEV site for tag removal.
Outcome: Affinity capture yield went from effectively zero to roughly 1.2 mg per liter of culture — enough to move into detergent screening. Net cost of the original error: two failed expression rounds (~5 weeks). Net cost of the topology check that would have prevented it: about ten minutes.
A Decision Tree for Reading Topology From Sequence
Construct Design Checklist
Before you order primers for a membrane protein, verify:
You predicted topology with a tool that models signal peptides, not a TM-only method
You confirmed which terminus is cytoplasmic before choosing tag location
Your affinity tag is on a cytoplasmic terminus or loop (in E. coli, for proper folding and Ni binding)
Any crystallization fusion (BRIL, T4L) is placed in a cytoplasmic loop long enough to host it
You checked the positive-inside charge balance by hand and it matches the predicted orientation
For channels/transporters, you flagged possible re-entrant loops and didn't truncate across one
No truncation boundary cuts inside a predicted TM helix (leaves a sticky hydrophobic stub)
If small/SMR-like, you ruled out dual topology from the literature
At least two predictors agree on the regions you're designing around
The Economics of Getting Topology Right
Approach | Time to Answer | Cost | What You Get |
|---|---|---|---|
Skip prediction, guess tag placement | 0 min | High (failed rounds) | Coin-flip orientation; weeks lost on bad constructs |
Hydropathy plot only | ~5 min | Free | Helix count, no orientation, no signal-peptide call |
Modern topology + signal-peptide predictor | ~10 min | Free | Count + orientation + signal handling, per-residue labels |
Two predictors + manual positive-inside check | ~30 min | Free | Same, plus a confidence read on disputed regions |
Experimental topology mapping (reporter fusions) | Weeks | High (cloning + assays) | Ground truth — reserve for the regions prediction can't settle |
ROI consideration: a single failed membrane-protein expression round burns 3–5 weeks and real reagent cost. The prediction that prevents it costs ten minutes. There is no membrane-protein project where reading topology first doesn't pay for itself.
Bottom Line
Read topology from sequence before you design the construct. Helix count comes from hydrophobicity; orientation comes from the positive-inside rule; signal peptide versus signal anchor must be resolved separately; and re-entrant loops, marginal helices, and dual topology are where even the best predictors quietly fail. Treat the prediction as a confidence-weighted hypothesis, cross-check two tools, and design your tags and truncations around what they agree on.
How Orbion Helps
Topology prediction shouldn't live in a separate web server disconnected from the rest of your construct design. In Orbion, it runs as part of the Characterization module, mapped directly onto your sequence and structure alongside the other tracks you need to make a construct decision.
Relevant Orbion features:
AstraUNFOLD — transmembrane topology, disorder, and amyloid in one pass: AstraUNFOLD predicts transmembrane helix topology using a six-label scheme at per-residue resolution, plus per-residue disorder probability and amyloid propensity. You read helix boundaries, in/out orientation, and disordered loop regions together — so a tag-safe cytoplasmic loop that's also low-disorder is obvious at a glance.
Characterization with Kyte–Doolittle structure coloring: the 3D viewer colors your predicted structure by hydrophobicity (Kyte–Doolittle), letting you see the membrane-embedded hydrophobic belt and cross-check it against the AstraUNFOLD topology track — the modern visual descendant of the hydropathy plot, mapped onto real coordinates.
Topology informs Design tag placement: the Design module assembles constructs (signal peptide → N-terminal tags → fusions → linker → target → C-terminal tags) and scores them. Knowing which terminus is cytoplasmic from AstraUNFOLD tells you which end to tag and which loops can host a solubility or stabilization fusion before you commit.
Topology feeds Bench expression-system choice: membrane type and TM-helix context flow into the Bench module's Context Wizard, where they inform the Rate of Ease difficulty score and the recommended expression system (E. coli, insect, mammalian, or cell-free) for a protein whose topology you now actually understand.
Upload a sequence and you get topology, disorder, hydrophobicity, and a construct-design workspace in one place — instead of stitching together three web servers and hoping their orientation calls agree.
References
Kyte J, Doolittle RF. (1982). A simple method for displaying the hydropathic character of a protein. Journal of Molecular Biology, 157(1):105-132. https://doi.org/10.1016/0022-2836(82)90515-0
von Heijne G. (1992). Membrane protein structure prediction: hydrophobicity analysis and the positive-inside rule. Journal of Molecular Biology, 225(2):487-494. https://doi.org/10.1016/0022-2836(92)90934-C
Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. Journal of Molecular Biology, 305(3):567-580. https://doi.org/10.1006/jmbi.2000.4315
Käll L, Krogh A, Sonnhammer ELL. (2004). A combined transmembrane topology and signal peptide prediction method. Journal of Molecular Biology, 338(5):1027-1036. https://doi.org/10.1016/j.jmb.2004.03.016
Hallgren J, Tsirigos KD, Pedersen MD, Almagro Armenteros JJ, Marcatili P, Nielsen H, Krogh A, Winther O. (2022). DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. bioRxiv. https://doi.org/10.1101/2022.04.08.487609
Teufel F, Almagro Armenteros JJ, Johansen AR, Gíslason MH, Pihl SI, Tsirigos KD, Winther O, Brunak S, von Heijne G, Nielsen H. (2022). SignalP 6.0 predicts all five types of signal peptides using protein language models. Nature Biotechnology, 40(7):1023-1025. https://doi.org/10.1038/s41587-021-01156-3
Viklund H, Granseth E, Elofsson A. (2006). Structural classification and prediction of reentrant regions in alpha-helical transmembrane proteins: application to complete genomes. Journal of Molecular Biology, 361(3):591-603. https://doi.org/10.1016/j.jmb.2006.06.037
Rapp M, Granseth E, Seppälä S, von Heijne G. (2006). Identification and evolution of dual-topology membrane proteins. Nature Structural & Molecular Biology, 13(2):112-116. https://doi.org/10.1038/nsmb1057
Almén MS, Nordström KJV, Fredriksson R, Schiöth HB. (2009). Mapping the human membrane proteome: a majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biology, 7:50. https://doi.org/10.1186/1741-7007-7-50
Käll L, Krogh A, Sonnhammer ELL. (2007). Advantages of combined transmembrane topology and signal peptide prediction — the Phobius web server. Nucleic Acids Research, 35(suppl_2):W429-W432. https://doi.org/10.1093/nar/gkm256
Book a 20-Minute Demo
Sign up free for unlimited Overview runs — summary, sequence-based analysis, homology search. For the full Characterization — PTMs, binding sites, stability variants, construct design — book a demo and we'll run your target live.
