Blog
Orbion Team
How Many Constructs Should You Screen in Parallel — and Which Ones?

You picked the construct that looked best on paper — full-length, an N-terminal His6, a TEV site — cloned it, expressed it, and got nothing in the soluble fraction. So you tried a C-terminal tag. Two weeks later, still nothing. Then you truncated the disordered N-terminus. Another two weeks. Three months in, you've tested four constructs one at a time and you're no closer to soluble protein than when you started.
The structural genomics community learned this lesson at scale a decade ago: when you commit to one construct and iterate serially, you're not optimizing — you're gambling slowly. A single eukaryotic construct expressed in E. coli succeeds less than 10% of the time. Test 9–15 constructs of the same target in parallel, and success rates climb to roughly 48% [1, 2].
Key Takeaways
Single constructs are a coin flip you'll lose: One expression construct for a eukaryotic target in E. coli succeeds <10% of the time. Parallel screening of 9–15 constructs per target raised the Structural Genomics Consortium's purification success to 48% across 1,269 targets [2].
Parallel beats serial on both math and time: Four serial rounds at two weeks each burns two months to test four options. The same four — plus eight more — fit in one parallel round at roughly the same wall-clock cost.
Diversity is the whole point: A construct set that varies only the tag wastes your wells. The variables that move the needle most are termini and domain boundaries, then fusion/solubility partner, then tag and vector. Spread your set across the high-leverage axes.
Vary-one-factor for diagnosis, combinatorial for coverage: Use a sparse one-factor-at-a-time (OFAT) design when you want to learn why something works; use a combinatorial block when you just need a hit and have the throughput to absorb it.
8–24 constructs is the practical sweet spot: Below 8 you under-sample the design space; above ~24 per target the marginal hit rate flattens and cost dominates. Pick the number from your target's risk profile, not a fixed rule.
Terminal truncations dominate crystallizable hits: At the SGC, 83% of solved structures came from truncated constructs, not full-length protein [2]. If you only screen tags, you're screening the wrong axis.

Why One-at-a-Time Loses
Serial construct optimization feels rigorous. You change one thing, you observe the result, you reason about the next change. The problem is the feedback loop length. Each cloning-expression-analysis cycle runs one to three weeks. A negative result tells you almost nothing actionable — "no soluble protein" is consistent with a bad tag, a bad boundary, a misfolded fusion, a toxic product, or codon problems all at once. You can't deconvolve a null.
So you guess at the next variable, change it, and wait again. Across structural genomics pipelines handling tens of thousands of targets, this serial instinct is exactly what high-throughput groups engineered away. The consensus "what to try first" review distilled from over 10,000 proteins is blunt about it: the single highest-yield decision is to clone and express multiple constructs of each target in parallel rather than perfecting one [1].
The core tension: serial screening optimizes your reasoning about one construct; parallel screening optimizes your probability of getting any construct to work. For an early-stage target where you don't yet know which variable is limiting, probability wins — because you don't have the information to reason well yet.

What the Structural Genomics Numbers Actually Show
The evidence for parallel multi-construct screening is unusually quantitative, because the structural genomics consortia ran it across thousands of targets and published the box scores.
The headline: ~5x over single constructs
A single eukaryotic construct in E. coli purifies to useful levels less than 10% of the time. The SGC's high-throughput pipeline — built around testing 9–15 constructs per target or domain in parallel — purified at least one construct for 614 of 1,269 targets (48%) [2]. That's roughly a five-fold lift, and it came almost entirely from breadth, not from any single clever trick.
Construct libraries double soluble expression
You don't need an industrial pipeline to see the effect. Cornvik and colleagues screened libraries that randomized the N-terminal translation start point across a panel of 32 mammalian proteins. Soluble, readily purifiable expression doubled from 34% to 68% [3]. Same proteins, same host, same week — the only change was screening many start points instead of betting on one.
Truncations carry the crystallizable hits
Where parallel screening pays off most is downstream. A systematic study of N- and C-terminal deletions across ~400 human targets nearly doubled soluble-expression success and produced a more than fourfold increase — from 15 to 65 — in targets yielding well-diffracting crystals [4]. At the SGC, 83% of solved structures came from truncated constructs; only 11% came from full-length protein [2]. The lesson isn't "truncate everything." It's that the boundary axis is where the diffraction-quality outcomes live, so your parallel set must sample it.
Tags and fusions are real but target-specific
Fusion partners don't perform uniformly. The same MBP or SUMO that rescues one target leaves the next insoluble — there is no universal best tag, only a best tag for this protein [5]. Parallel fusion-screening systems exist precisely because the only way to know is to test several side by side [5]. That target-specificity is the argument for diversity, not against tags: you screen a spread because you can't predict which member of the spread your protein prefers.
Note: this article is about the parallel screening strategy — how many constructs and which axes. For how to choose specific domain boundaries, see our construct-boundary series; for picking a specific fusion partner, see the fusion-partner selection guide. Here we assume you'll vary those axes and focus on how to lay out the round.

How Many Constructs? Sizing the Round
There's no universal number, but the design space and the cost curve bracket it tightly.
The under-sampling floor
Below about 8 constructs, you can't meaningfully cover more than one or two axes. If you only have budget for four wells, you'll spend them all on tags or all on boundaries and learn nothing about the other axes. The structural genomics groups that hit ~48% were testing 9–15 per target for a reason: that's roughly the number needed to put a few truncations, a couple of fusion options, and a tag-placement choice into the same round [2, 6].
The diminishing-returns ceiling
Hit rate is not linear in construct count. The first handful of well-chosen, diverse constructs captures most of the achievable success. Doubling from 12 to 24 adds coverage but a smaller marginal lift; going past ~24 per target mostly adds cost and analysis burden. The exception is high-throughput combinatorial screening, where the marginal cost per construct is low enough that you can afford to over-sample — more on that below.
Match the count to target risk
Target profile | Suggested round size | Why |
|---|---|---|
Well-behaved bacterial cytosolic protein | 4–8 | High base rate; a few boundary/tag variants suffice |
Eukaryotic soluble protein, novel | 12–18 | Low base rate; sample boundaries + fusions + tag placement |
Multidomain or disordered-region-heavy | 16–24 | Boundary uncertainty is the dominant risk; over-sample termini |
Membrane protein / known-hard | 16–24+ | Low base rate, many variables; combinatorial often justified |
The number falls out of how much boundary uncertainty you carry and how low your base success rate is. A novel eukaryotic multidomain protein deserves a wide round; a close homolog of a protein you've crystallized before does not.
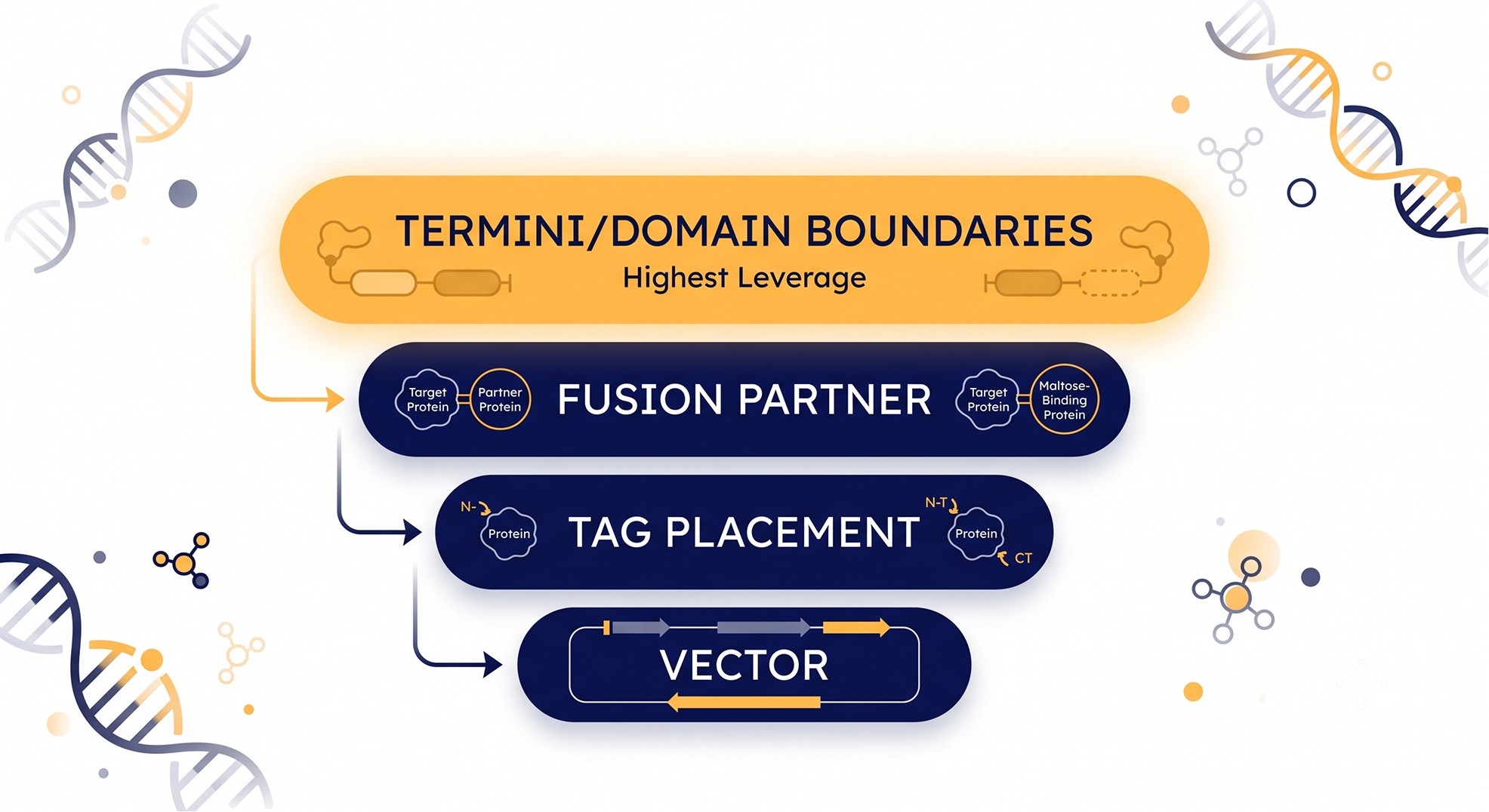
Which Constructs? Designing a Diverse, Informative Set
Twelve constructs that differ only in tag identity is not a parallel screen — it's one experiment cloned twelve times. The value of a parallel round is proportional to how much of the outcome-relevant design space it covers. Rank your axes by leverage and spend your wells accordingly.
The leverage ranking
Termini / domain boundaries — highest leverage. Drives both soluble expression and, critically, crystallizability [2, 4]. Always sample several.
Fusion / solubility partner — high leverage, target-specific. MBP, SUMO, GST, TrxA, NusA behave differently per target [5]. Include 2–3 options for hard targets.
Tag placement and identity — moderate leverage. N- vs C-terminal placement, His6 vs Strep, cleavable vs not. Cheap to vary.
Vector / expression context — lower per-construct leverage but matters at the margins; promoter strength, RBS, host strain.
OFAT vs combinatorial
There are two ways to lay out the round, and they answer different questions.
Vary one factor at a time (OFAT) gives you a sparse, interpretable set. Fix a sensible baseline construct, then change exactly one thing per construct: one truncation, one fusion swap, one tag move. With ~12 constructs you cover a baseline plus a handful of single-variable changes. When a construct works, you know which change caused it — the round doubles as a diagnostic. Use OFAT when you want transferable knowledge for the next target in a family.
Combinatorial crosses your axis options multiplicatively: 3 boundaries × 3 fusions × 2 tag placements = 18 constructs covering every combination. You stop reasoning and start saturating. Combinatorial finds interaction effects that OFAT misses — the truncation that only works with SUMO, for instance — but a single hit doesn't tell you which factor mattered. Use combinatorial when you need a working construct fast, the target is hard, and your cloning throughput can absorb the count.
The honest tradeoff: OFAT is information-efficient but coverage-poor; combinatorial is coverage-rich but information-poor. Most well-run rounds are a hybrid — a small OFAT spine for interpretability, padded with a few combinatorial crosses on the highest-leverage axes.

Case Study: Rescuing a Stalled Eukaryotic Target
Problem: A team had spent three months on a 47 kDa human signaling protein. They had cloned full-length protein with an N-terminal His6-TEV, gotten no soluble expression, then serially tried a C-terminal His6 (nothing), an MBP fusion (soluble but wouldn't cleave cleanly), and a Strep-tag (nothing). Four constructs, three months, no usable material. Each null result was uninterpretable because too many variables differed between attempts.
Analysis: Disorder prediction flagged a 38-residue intrinsically disordered N-terminal extension and a flexible 22-residue C-terminal tail — both untested as truncation points. The protein had two predicted domains with a flexible linker, suggesting a single-domain construct might behave better than full-length. The fusion result (soluble with MBP) was a strong signal that the folding was rescuable; the limiting variable was almost certainly boundaries, not tag chemistry.
Solution: Instead of a fifth serial guess, the team laid out one parallel round of 16 constructs: 4 N-terminal start points (full, Δ38, Δ20, domain-1-start) × 2 C-terminal ends (full, Δ22) × 2 fusion contexts (His6 only, SUMO). Combinatorial on boundaries, two fusion options, one cloning campaign using ligation-independent cloning so all 16 went in together.
Outcome: 5 of 16 constructs gave soluble expression — a 31% hit rate within the round. The best performer was Δ38/Δ22 with a cleavable SUMO fusion; it expressed at 8 mg/L, cleaved cleanly, and ran as a monodisperse single SEC peak. Total time from design to soluble protein: 18 days — shorter than any single one of their prior serial cycles, and it produced a crystallizable construct the serial approach had never sampled because it never tested two truncations together.
Practical Checklist
Before you commit a parallel construct round, verify:
You've ranked your axes by leverage — boundaries and fusions before tags
Termini are sampled — at least 2–3 truncation variants for any novel eukaryotic target
The round size matches target risk — 4–8 for easy, 12–24 for hard/novel
At least one fusion option is included for low-base-rate targets
A clear baseline construct exists so OFAT changes stay interpretable
You chose OFAT vs combinatorial deliberately based on whether you need knowledge or a hit
Cloning method scales to the count (LIC / Gibson / parallel kit, not one-by-one restriction cloning)
Vector compatibility is checked — your chosen tags/fusions actually exist in vectors you can source
A go/no-go readout is defined — small-scale solubility (gel, CE, or dot-blot) before scale-up
The Economics
The cost case for parallel screening is not subtle once you put serial and parallel on the same axes. Assume a two-week cycle per serial attempt and a per-construct marginal cost dominated by cloning reagents and a small-scale expression check.
Strategy | Constructs | Wall-clock time | Relative cost | Expected success |
|---|---|---|---|---|
Single construct, full-length | 1 | 2 weeks | 1x | <10% (eukaryotic in E. coli) [1] |
Serial, 4 attempts | 4 | ~8 weeks | ~4x labor | Modest; uninterpretable nulls |
Parallel OFAT round | 8–14 | ~3 weeks | ~2–3x reagents, ~1x time | ~30–48% [2, 4] |
Parallel combinatorial round | 16–24+ | ~3–4 weeks | ~3–5x reagents, ~1x time | ~30–48%+, finds interactions |
ROI consideration: The expensive resource in protein production is rarely reagents — it's calendar time and scientist attention. Serial screening's hidden cost is that it spends weeks to buy one low-probability draw, and the nulls don't even tell you what to try next. A parallel round spends a multiple of the reagent cost to buy 8–24 simultaneous draws at the same wall-clock cost, and a diverse round returns interpretable information whether it hits or not. When a stalled target blocks a downstream campaign worth months of team time, the reagent delta is rounding error.
Bottom Line
Stop optimizing one construct and start screening many. For any novel or eukaryotic target, lay out a single parallel round of 8–24 diverse constructs — weighted toward terminal/boundary variants, with at least one fusion option — instead of iterating serially. The structural genomics evidence is unambiguous: breadth raises success from a coin-flip-you-lose to roughly even odds, at the same wall-clock cost.
How Orbion Helps
Designing a diverse, well-balanced construct round by hand is tedious and error-prone — you're juggling truncation boundaries, tag chemistries, fusion partners, vector compatibility, and codon optimization across a dozen-plus variants, then trying to predict which ones are worth the bench time. Orbion's Design module is built for exactly this layout problem.
Relevant Orbion features:
AI generation of 5 diverse construct strategies: From your sequence, the Design module's AI generation wizard proposes five distinct construct strategies — varying boundaries, tags, and fusions — each with a rationale and confidence score, so your parallel round starts diverse by construction instead of by guesswork.
Combinatorial mode with live variant counter: Cross your tag, linker, and fusion options to generate up to 5,000 variants, with a live counter showing the combinatorial size as you build — turning "which combinations should I clone?" into a deliberate, bounded design rather than a serial slog.
Composite scoring before you clone: Every construct is automatically scored on solubility, disorder, aggregation, ΔTm (via AstraDTM), and ΔΔG (via AstraDDG), combined into a sortable composite score against a pinned wild-type reference — so you can rank a 24-construct set and shortlist the most promising before committing a single well.
Vector-library compatibility matching: The Design module matches each construct against your organization's vector library and flags exact, partial, and no-match counts — catching the "this tag combination doesn't exist in any vector I can source" problem at the design stage, not after you've ordered primers.
One-click handoff to Bench: Shortlist your round, then generate construct-aware experimental protocols for the winners with a single "Create Bench Design" — the assembled protein and codon-optimized DNA sequences carry straight through to expression and purification planning.
Instead of testing four constructs over three months and learning nothing from the nulls, you can design, score, and shortlist a diverse 16-construct round in an afternoon — then clone the ones the model says are worth your bench time.
References
Structural Genomics Consortium, China Structural Genomics Consortium, Northeast Structural Genomics Consortium, et al. (2008). Protein production and purification. Nature Methods, 5(2):135–146. Link
Savitsky P, Bray J, Cooper CDO, et al. (2010). High-throughput production of human proteins for crystallization: The SGC experience. Journal of Structural Biology, 172(1):3–13. Link
Cornvik T, Dahlroth SL, Magnusdottir A, et al. (2006). An efficient and generic strategy for producing soluble human proteins and domains in E. coli by screening construct libraries. Proteins, 65(2):266–273. Link
Gräslund S, Sagemark J, Berglund H, et al. (2008). The use of systematic N- and C-terminal deletions to promote production and structural studies of recombinant proteins. Protein Expression and Purification, 58(2):210–221. Link
Esposito D, Chatterjee DK. (2006). Enhancement of soluble protein expression through the use of fusion tags. Current Opinion in Biotechnology, 17(4):353–358. Link
Rasia RM, Noirclerc-Savoye M, Bologna NG, et al. (2009). Parallel screening and optimization of protein constructs for structural studies. Protein Science, 18(2):434–439. Link
Book a 20-Minute Demo
Sign up free for unlimited Overview runs — summary, sequence-based analysis, homology search. For the full Characterization — PTMs, binding sites, stability variants, construct design — book a demo and we'll run your target live.
