Blog
Orbion Team
The Solubility Tag Paradox: Why Your Protein Precipitates After Tag Cleavage

Your MBP-fusion was crystal clear at 10 mg/mL. The SEC trace was a textbook symmetric peak. You added TEV protease at a 1:50 molar ratio and left it overnight at 4°C. By morning, the tube looked like skim milk—a dense white precipitate had crashed out of solution, and the SDS-PAGE confirmed your worst fear: the cleaved MBP was still in the supernatant, but your target protein was almost entirely in the pellet. This is the solubility tag paradox, and it is one of the most demoralizing failure modes in recombinant protein production.
The fusion partner was never solubilizing your protein in the thermodynamic sense. It was masking an aggregation problem that returns the instant the mask comes off.
Key Takeaways
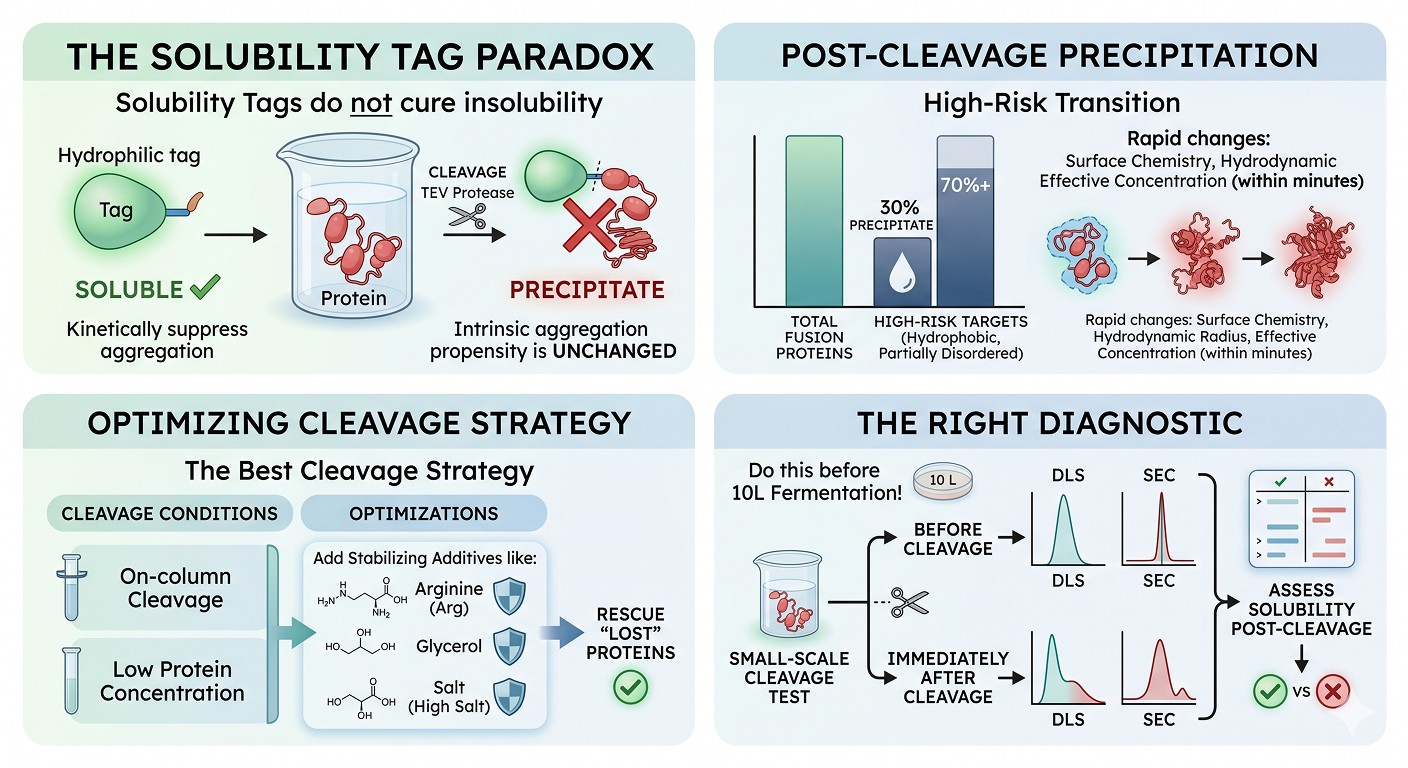
Solubility tags do not "cure" insolubility—they kinetically suppress aggregation by adding a large hydrophilic surface, but the passenger's intrinsic aggregation propensity is unchanged
Post-cleavage precipitation affects an estimated 30% of fusion proteins, and the rate is far higher for hydrophobic, aggregation-prone, or partially disordered targets
The cleavage event is the highest-risk transition in the entire purification: surface chemistry, hydrodynamic radius, and effective concentration all change within minutes
On-column cleavage at low concentration with stabilizing additives (arginine, glycerol, salt) rescues a large fraction of "lost" proteins
The right diagnostic is DLS or SEC before and immediately after a small-scale cleavage test—do this before committing 10 L of fermentation to a fusion strategy that will fail at the last step
What MBP, SUMO, and GST Are Actually Doing
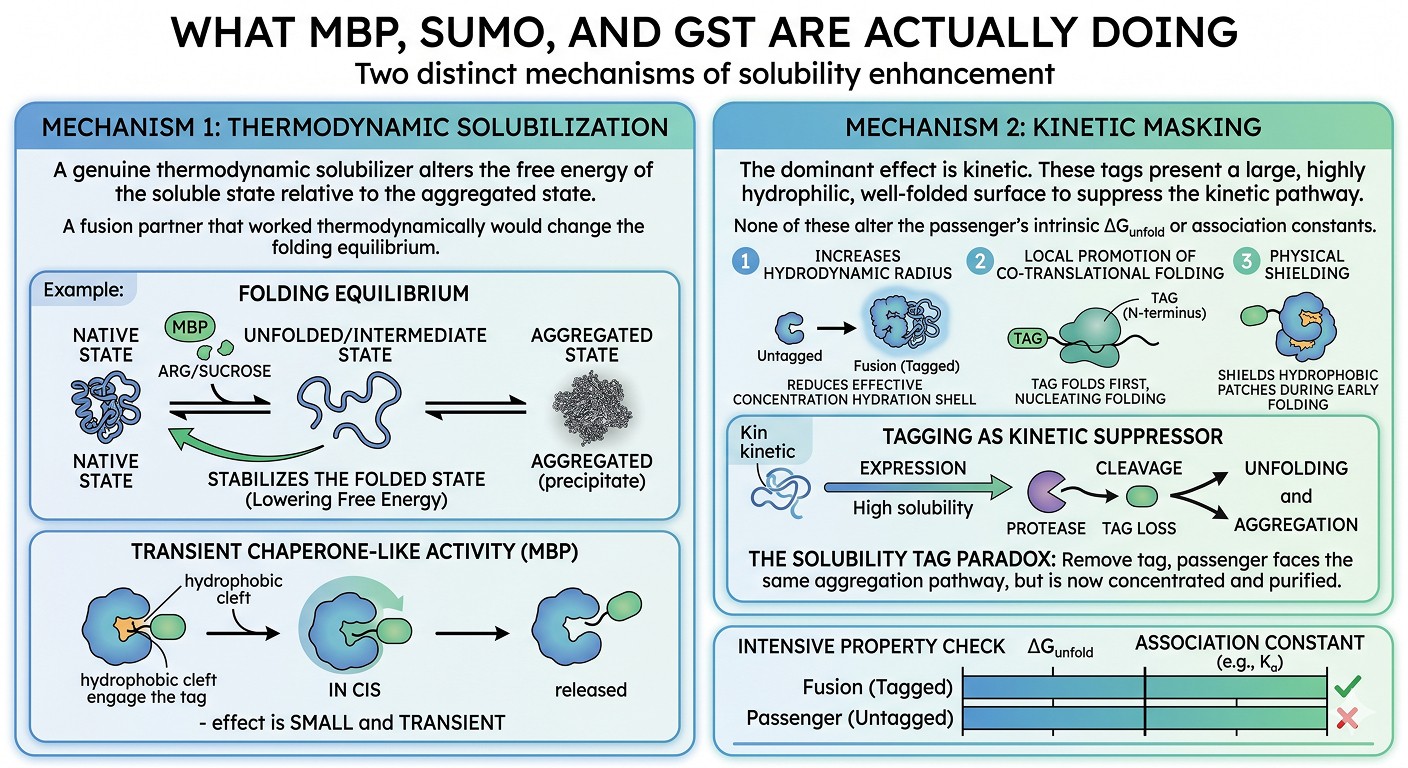
Before diagnosing why cleavage causes precipitation, it helps to be precise about the mechanism of solubility enhancement. The popular textbook description—"the tag makes the protein soluble"—conflates two very different physical effects, and the distinction is what determines whether your protein will survive cleavage.
Mechanism 1: Thermodynamic Solubilization
A genuine thermodynamic solubilizer alters the free energy of the soluble state relative to the aggregated state. The classic example is the addition of arginine or sucrose to a buffer—these excipients preferentially exclude from the protein surface and stabilize the more compact (less aggregation-prone) folded state.
A fusion partner that worked thermodynamically would change the folding equilibrium of the passenger. There is some evidence MBP does this for certain passengers via a chaperone-like activity that has been observed in vitro (Kapust & Waugh, 1999). MBP's binding cleft has weak affinity for hydrophobic peptides, and in cis (within a fusion) this can transiently engage hydrophobic patches on the passenger during folding.
But this effect is small and transient. Once the passenger has folded (or misfolded), MBP releases it, and the equilibrium between native and aggregated states is governed by the passenger's intrinsic surface chemistry—not by MBP.
Mechanism 2: Kinetic Masking
The dominant effect of MBP, SUMO, GST, and Trx is kinetic. Each of these tags presents a large, highly hydrophilic, well-folded surface that does several things at once:
Increases the hydrodynamic radius of the fusion, reducing the effective concentration of aggregation-prone surfaces per unit volume
Provides a folding nucleus that folds first and locally promotes co-translational folding of the passenger (the "folding-from-the-N-terminus" effect)
Physically shields hydrophobic patches on the passenger from intermolecular contacts during the critical early-folding window
Improves expression-level solubility by accelerating folding faster than aggregation can occur
None of these alter the passenger's intrinsic ΔG_unfold or its association constants for self-association. They simply suppress the kinetic pathway to aggregation during expression and early purification (Esposito & Chatterjee, 2006).
This is why the paradox exists. Strip the tag away, and the passenger now faces the same aggregation pathway it would have faced without the tag—except now you have purified, concentrated, pristine substrate for that pathway, and the protease is sitting in the tube to keep things moving.
What Actually Changes at the Moment of Cleavage
Cleavage is not a clean "tag off, protein still happy" event. Five things change simultaneously, and any one of them can trigger aggregation.
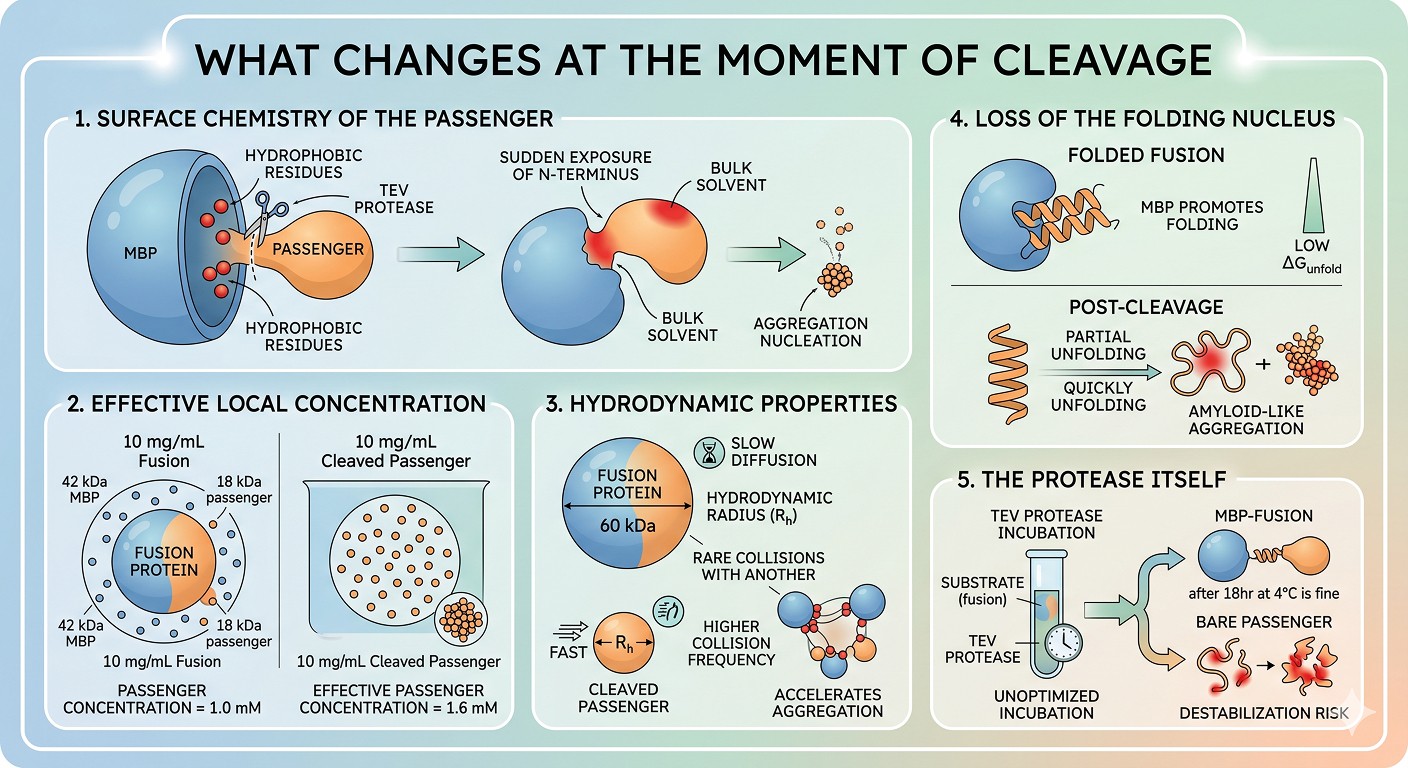
1. Surface Chemistry of the Passenger
The most underappreciated effect: when MBP is fused N-terminally to a passenger, the residues near the passenger's N-terminus are in contact with MBP. If those residues are hydrophobic—which they often are, because real protein N-termini frequently begin with a hydrophobic α-helix or β-strand—they were buried against MBP during the soluble fusion state.
The moment TEV cleaves, those residues are suddenly exposed to bulk solvent. The "newly born" N-terminus may include hydrophobic side chains that were never solvent-accessible in the fusion. If those side chains form an aggregation-promoting patch, you will see immediate nucleation.
2. Effective Local Concentration
A fusion protein at 10 mg/mL is, in molar terms, a much lower concentration than the cleaved passenger at 10 mg/mL—because the fusion includes 42 kDa of MBP. A 60 kDa MBP-passenger fusion at 10 mg/mL contains only about 60% of the passenger by mass. After cleavage and complete MBP removal, that same mass of passenger represents a higher molar concentration of the aggregation-prone species.
For aggregation that proceeds via second-order kinetics (the typical scenario for nucleation-dependent aggregation), doubling the effective concentration of the passenger more than doubles the aggregation rate.
3. Hydrodynamic Properties
MBP-passenger has a much larger hydrodynamic radius than the bare passenger. Larger objects diffuse more slowly and encounter each other less often. After cleavage, the smaller, more mobile passenger has a higher collision frequency. For nucleation-limited aggregation, this is exactly the wrong direction.
4. Loss of the Folding Nucleus
If the passenger requires the fusion partner not just for folding but for staying folded, removal of the tag can destabilize the native state. Marginally stable proteins—those with low ΔG_unfold—may partially unfold after tag removal. Partially unfolded states expose hydrophobic cores and are the universal precursor to amyloid-like aggregation (Bondos & Bicknell, 2003).
5. The Protease Itself
TEV protease at typical working concentrations adds a small amount of protein to the mixture, but more importantly it has been incubated with the substrate at temperatures and times that may not have been optimized for the passenger's stability. An 18-hour incubation at 4°C is fine for MBP-fusion. It may not be fine for the bare passenger.
Why MBP Looks Different From SUMO Looks Different From GST
The post-cleavage behavior of a passenger depends critically on which tag was used. They are not interchangeable.
MBP: Maximum Suppression, Maximum Bounce-Back
MBP at 42.5 kDa is the largest of the standard solubility tags and the most effective at kinetic masking (Fox & Waugh, 2003). This is also why MBP-fusions are most likely to exhibit dramatic post-cleavage precipitation: the gap between "what the fusion looks like" and "what the bare passenger looks like" is the largest.
A protein that is genuinely soluble on its own merits will be soluble as an MBP fusion too. But a protein that needs significant kinetic masking will sail through the MBP-fusion stage and then crash hard at cleavage. MBP is the tag most likely to give you a false sense of security.
SUMO: Smaller Mask, Smaller Surprise
SUMO at ~11 kDa provides less kinetic masking than MBP. The advantage is that proteins that survive as SUMO fusions are usually genuinely closer to soluble on their own—SUMO is more diagnostic. The Ulp1/SENP cleavage also leaves a native N-terminus, which means there is no extra-residue surface change to worry about (Butt et al., 2005).
When SUMO-fused proteins do precipitate after cleavage, the precipitation tends to be more diagnostic of a real underlying problem (marginally stable fold, disordered region, surface hydrophobic patch) rather than the simple kinetic transition from "MBP-shielded" to "exposed."
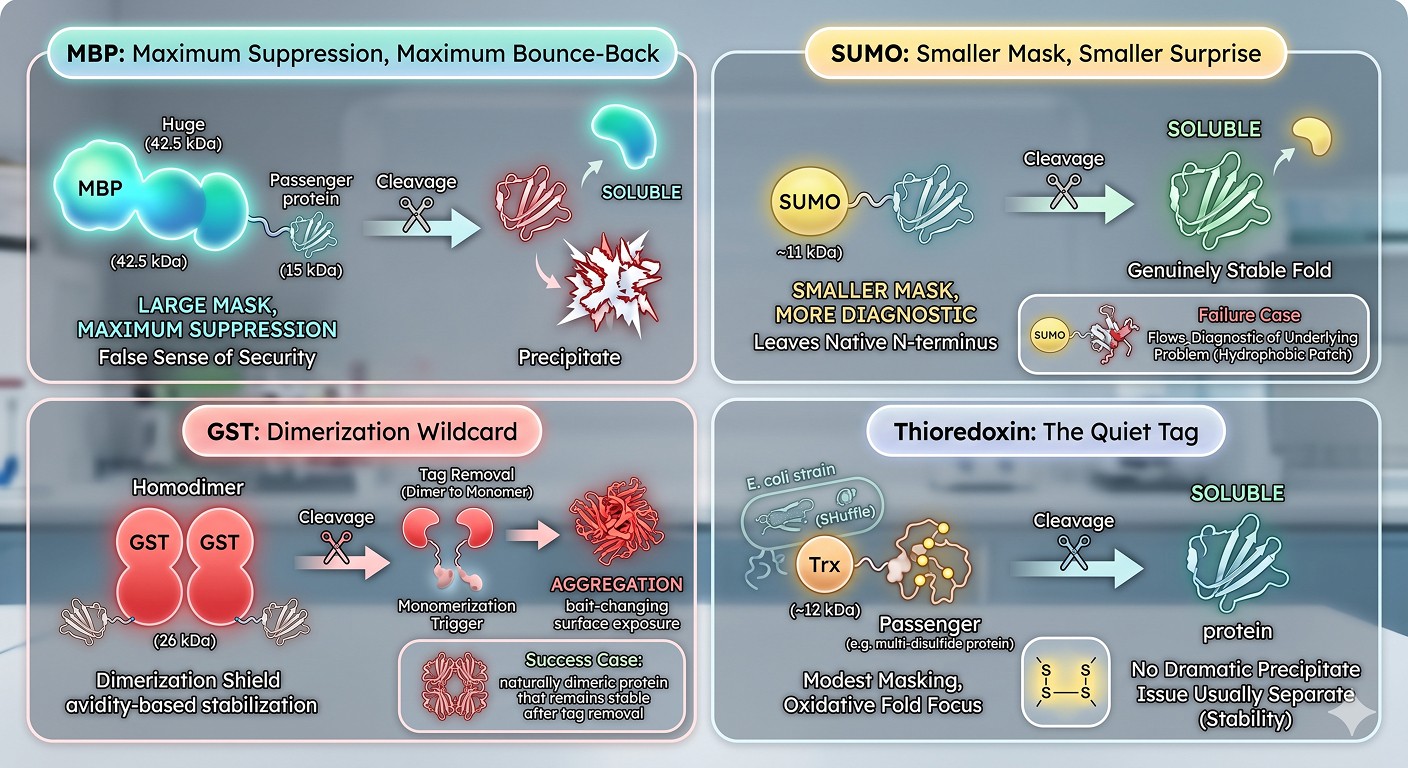
GST: Dimerization Wildcard
GST at ~26 kDa is a homodimer. GST-fusions therefore present the passenger as part of a dimer. Some passengers tolerate this; for some, the forced dimerization causes problems even before cleavage. After cleavage of GST, two things happen at once:
The kinetic mask is removed
The artificial dimerization is removed
For passengers that were partially soluble as GST-fusion dimers because of avidity-based stabilization (intermolecular contacts between two passenger copies bridged by GST dimerization), monomerization itself can trigger aggregation. PreScission/3C cleavage of GST is a multi-step transition: dimer → monomer + bait-changing surface exposure.
Thioredoxin: The Quiet Tag
Trx at ~12 kDa provides modest masking but typically does not produce dramatic post-cleavage precipitation. This is partly because Trx is small enough that the kinetic gap is small, and partly because Trx is most commonly used for proteins with disulfide bonds in the SHuffle strain—where the underlying problem is oxidative folding, not surface hydrophobicity (LaVallie et al., 1993). When Trx-fusion proteins precipitate after cleavage, the cause is usually a separate stability issue.
Post-Cleavage Success Rates by Tag
The following table summarizes typical observations from systematic surveys and structural genomics pipelines. The numbers are approximate and depend heavily on the passenger class.
Fusion Tag | Soluble Fusion Rate | Soluble After Cleavage Rate | "Cleavage Loss" |
|---|---|---|---|
MBP | ~75–80% | ~50–55% | ~25–30 pp |
SUMO | ~60–65% | ~50–55% | ~10–15 pp |
GST | ~45–55% | ~35–40% | ~10–15 pp |
Thioredoxin | ~50–60% | ~45–55% | ~5–10 pp |
NusA | ~70–75% | ~40–45% | ~25–30 pp |
GB1 | ~40–50% | ~40–45% | ~5 pp |
The pattern is consistent with the kinetic-masking hypothesis: the larger and more effective the mask, the larger the gap between fusion solubility and cleaved solubility. MBP and NusA—the two strongest masks—produce the most dramatic post-cleavage losses (Costa et al., 2014).
Aggregation Kinetics After Cleavage: What the Curves Tell You
The timescale of post-cleavage precipitation is itself diagnostic. Different timescales implicate different mechanisms, and each suggests a different intervention.
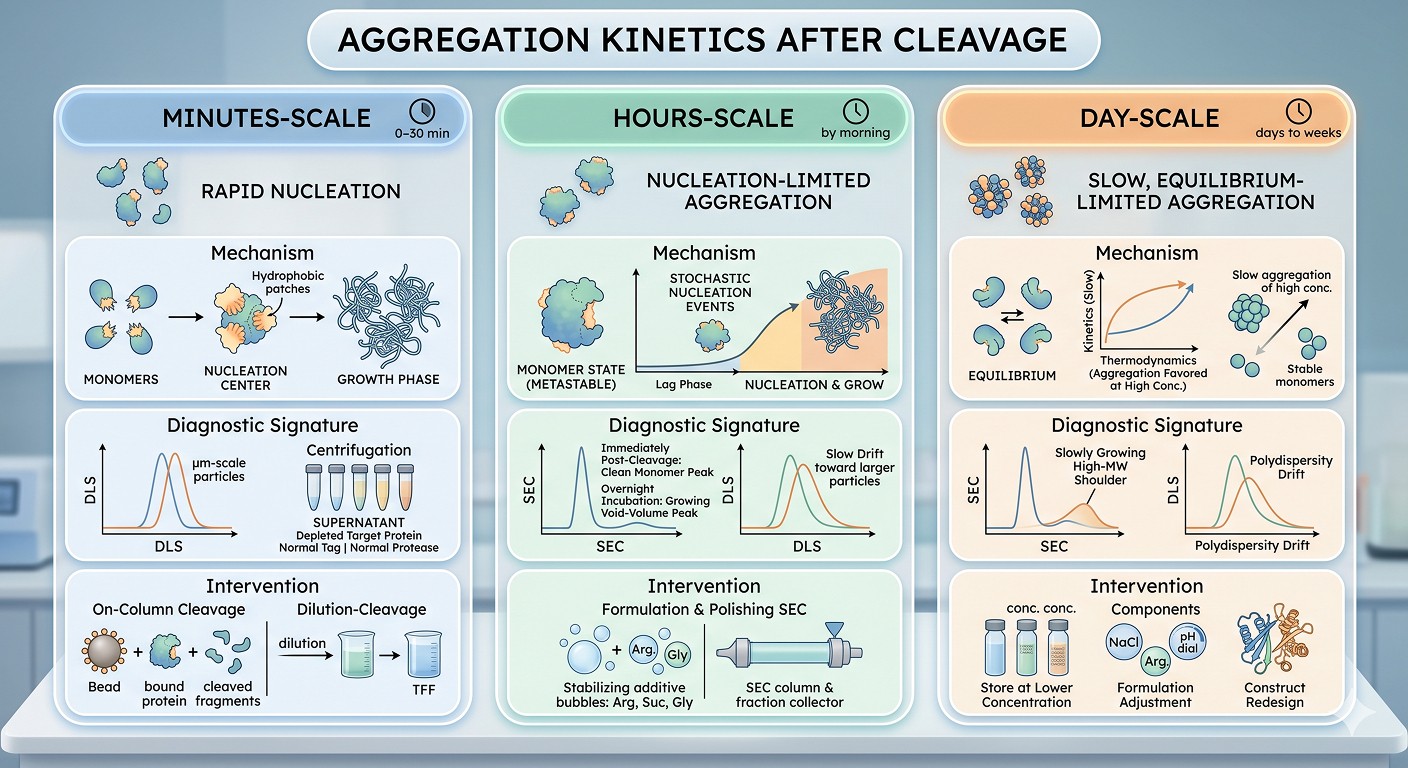
Minutes-Scale Precipitation (Visible Cloudiness Within 5–30 min)
What is happening: Rapid nucleation. The cleaved passenger has a strong hydrophobic patch (or several) on its surface, and intermolecular contacts form on a diffusion-limited timescale. Once nuclei form, growth proceeds rapidly because the population of aggregation-competent monomers is large.
Diagnostic signature: DLS shows the appearance of large (μm-scale) particles within minutes. The supernatant after centrifugation contains depleted target protein but normal levels of cleaved tag and protease.
Intervention: This protein cannot be cleaved in batch at working concentrations. Use on-column cleavage with the substrate at low density on the resin, or use dilution-cleavage (dilute 10x, cleave, reconcentrate via tangential flow at 4°C).
Hours-Scale Precipitation (Visible by Morning, Not at 1 Hour)
What is happening: Nucleation-limited aggregation with a meaningful lag phase. The protein is metastable in solution—soluble for a window, but will eventually aggregate. The lag phase reflects the time required for stochastic nucleation events.
Diagnostic signature: SEC immediately post-cleavage shows a clean monomer peak; SEC after overnight incubation shows a growing void-volume peak. DLS shows a slow drift toward larger particles.
Intervention: Move to formulation immediately after cleavage. Add stabilizing excipients (arginine, sucrose, glycerol), perform polishing SEC at low temperature within 2 hours of cleavage, and concentrate only after polishing—not before.
Day-Scale Precipitation (Days to Weeks, Concentration-Dependent)
What is happening: Slow, equilibrium-limited aggregation. The protein is genuinely close to soluble, but at high concentrations the aggregated state is thermodynamically favored. This is often seen with marginally stable proteins or proteins with a "sticky" surface but no dominant aggregation patch.
Diagnostic signature: SEC shows a slowly growing high-MW shoulder over days. DLS may show a polydispersity drift before any obvious cloudiness.
Intervention: Store at lower concentration than your "preferred" working concentration. Reformulate buffer (raise NaCl, add arginine, consider a different pH). Consider construct redesign to remove the offending surface.
Diagnostics: Catching the Problem Before You Scale Up
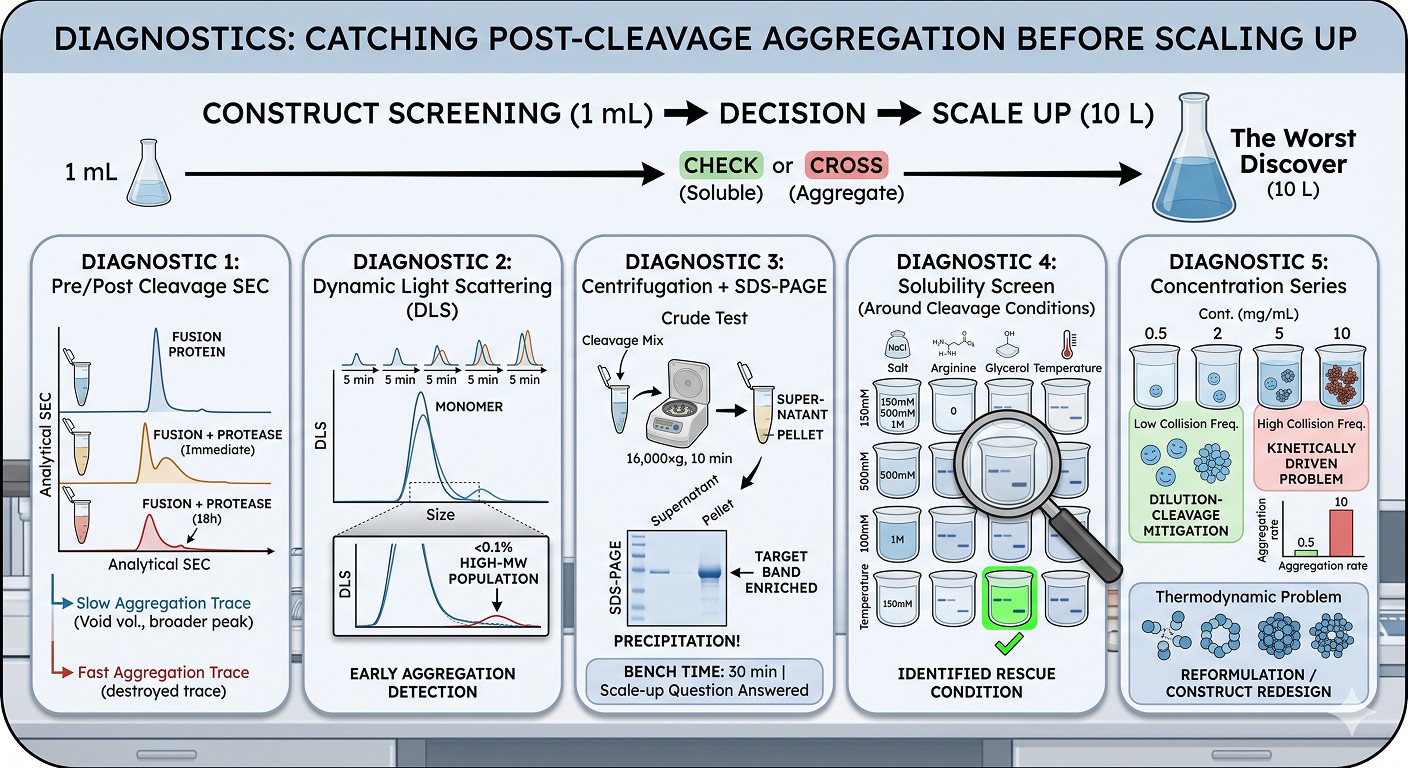
The worst version of the solubility tag paradox is the one you discover at 10 L scale. The right time to discover it is during construct screening at 1 mL scale.
Diagnostic 1: Pre/Post Cleavage SEC
Run analytical SEC on three samples:
Fusion protein (post-IMAC, post-buffer-exchange)
Fusion + protease, immediately after cleavage is complete
Fusion + protease, 18 hours post-cleavage at 4°C
A clean, monodisperse peak in sample 1, a clean peak in sample 2, and a degraded peak (void volume shoulder, broadened main peak) in sample 3 is the classic signature of slow post-cleavage aggregation. A clean peak in 1 and a destroyed trace in 2 indicates fast post-cleavage aggregation.
Diagnostic 2: Dynamic Light Scattering
DLS is the most sensitive method for early-stage aggregation. It can detect aggregates at <0.1% of total protein. Run DLS at 5-minute intervals after adding protease to your fusion. The appearance of a high-MW population—even one that does not yet visibly cloud the sample—indicates that aggregation is occurring and a fix is needed.
Diagnostic 3: Centrifugation + SDS-PAGE
The crudest but most decisive test: cleave a small sample, centrifuge at 16,000×g for 10 minutes, and run supernatant and pellet on SDS-PAGE. If your target band is enriched in the pellet relative to the cleaved tag, you have post-cleavage precipitation.
This is the test to run before scaling up. It costs 30 minutes of bench time and answers the only question that matters: "Will I have soluble protein at the end of this prep?"
Diagnostic 4: Solubility Screen Around the Cleavage Conditions
The conditions used during cleavage are often not optimized for the bare passenger. Test cleavage under varied conditions:
Salt: 150 mM vs 500 mM vs 1 M NaCl
Arginine: 0 vs 100 mM vs 500 mM
Glycerol: 0 vs 10% vs 20%
Temperature: 4°C vs 16°C vs 25°C
pH: ±1 unit around your standard buffer
A 4×4 matrix of conditions tested with the centrifugation/SDS-PAGE diagnostic above will frequently identify a "rescue" condition where cleavage proceeds and the passenger remains soluble.
Diagnostic 5: Concentration Series
Cleave at multiple starting concentrations (e.g., 0.5, 2, 5, 10 mg/mL of fusion). If precipitation depends strongly on concentration, the problem is kinetically driven by molar collision frequency and can be mitigated by dilution-cleavage. If precipitation occurs even at low concentration, the problem is thermodynamic and requires reformulation or construct redesign.
Rescue Strategies: Eight Approaches That Actually Work
When you have identified post-cleavage precipitation, the following interventions—ranked roughly by ease of implementation—rescue most proteins.
Strategy 1: On-Column Cleavage at Low Density
The simplest fix. Instead of eluting your fusion from IMAC and then cleaving in batch:
Load fusion onto IMAC resin
Wash thoroughly
Add protease in elution buffer (or wash buffer) directly to the column
Incubate the column for 1–4 hours
Elute—the cleaved passenger comes off the column, the His-tagged MBP (or His-SUMO) remains bound
The passenger is exposed to the lowest possible concentration during the cleavage event, and the diffusion-limited collisions that drive nucleation are suppressed. This is often the difference between a soluble final product and a precipitated mess.
Strategy 2: Dilution at the Moment of Cleavage
If on-column cleavage is impractical (large prep volumes, low resin capacity), dilute the fusion 5–10x with cleavage buffer immediately before adding protease. The reduced concentration suppresses second-order aggregation kinetics. Concentrate after polishing SEC, not before.
Strategy 3: Arginine in the Cleavage Buffer
Arginine at 100–500 mM is a remarkable aggregation suppressor (Arakawa et al., 2007). Its mechanism is not fully understood, but it appears to coat hydrophobic surfaces without destabilizing native folds. Many proteins that aggregate immediately after cleavage in standard buffer will stay in solution if arginine is present during cleavage.
Caveat: arginine interferes with some downstream applications. It must be removed (typically by dialysis or rapid buffer exchange) before applications sensitive to its presence.
Strategy 4: Increase Ionic Strength
For proteins whose post-cleavage aggregation is driven by electrostatic surface patches, raising NaCl to 300–500 mM or adding 50–100 mM Na₂SO₄ can suppress aggregation. Test this systematically in the cleavage condition screen.
Strategy 5: Co-Cleavage Stabilizers
Glycerol (10–20%), sucrose (5–10%), or trehalose (5–10%) are preferentially excluded osmolytes that stabilize folded states. Adding any of these to the cleavage buffer suppresses unfolding-mediated aggregation.
Strategy 6: Switch Tags
If MBP-cleavage precipitates your protein, try SUMO. The smaller kinetic gap means SUMO-fusion behavior is more diagnostic, and Ulp1 cleavage leaves a native N-terminus (avoiding the extra-Ser problem from TEV).
Conversely, if SUMO-cleavage precipitates, MBP may give a window of higher post-cleavage solubility if combined with on-column cleavage—the larger kinetic mask buys time during the cleavage event itself.
Strategy 7: Dual Tags
A His₆-MBP-TEV-(passenger)-SUMO-Strep construct is more work to clone but offers two independent rescue strategies: cleave MBP first (with TEV) to free the N-terminus while SUMO continues to mask C-terminal aggregation, then cleave SUMO separately. This is the heavy-machinery approach for very difficult proteins.
Strategy 8: Construct Redesign
If all kinetic interventions fail, the protein needs to be redesigned. Common interventions:
Trim disordered termini. A 30-residue N-terminal IDR is often the aggregation driver. Identifying and removing it via AstraUNFOLD-style disorder analysis can dramatically improve post-cleavage solubility.
Surface mutagenesis. Identify exposed hydrophobic residues that are not part of any binding interface and mutate to polar residues (Leu→Lys, Phe→Glu).
Loop deletion or shortening. Long surface-exposed loops are common aggregation hotspots.
Domain isolation. Express individual stable domains rather than a multi-domain construct that has a problematic interdomain linker.
The Cleavage Condition Optimization Matrix
A practical screening matrix for a difficult passenger, run at small scale before scaling up:
Variable | Range to Test | Typical "Rescue" Setting |
|---|---|---|
Starting fusion concentration | 0.5–10 mg/mL | 1–2 mg/mL |
NaCl | 150–1000 mM | 300–500 mM |
Arginine | 0–500 mM | 100–250 mM |
Glycerol | 0–20% | 10% |
Temperature | 4°C–25°C | 4°C |
pH | ±1 from working pH | ½ unit toward higher I |
Protease:substrate ratio | 1:20–1:200 | 1:50 |
Cleavage time | 2–18 h | Shortest that gives >90% cleavage |
On-column vs in-solution | Both | On-column for difficult proteins |
Reducing agent | 1–5 mM DTT or TCEP | Match the passenger's redox biology |
The goal is not to optimize every parameter—it is to identify the one or two variables that dominate the rescue, and to set those decisively.
A Worked Example: The Marginally Stable Kinase Domain
A representative case from the protein production literature: a 35 kDa kinase catalytic domain expresses well as His₆-MBP-TEV-kinase in E. coli, yielding ~40 mg/L of soluble fusion at 4 mg/mL after IMAC. The fusion is monodisperse by SEC, and TEV digestion is complete within 4 hours at 4°C.
After TEV cleavage and reverse IMAC to remove His₆-MBP:
The eluate is visibly cloudy within 30 minutes
SDS-PAGE of the supernatant shows ~20% of the expected target band
The pellet contains the rest
The diagnostic workflow proceeds:
DLS during cleavage: Shows rapid appearance of >1 μm particles starting at ~15 minutes
Concentration series: At 0.5 mg/mL, cleavage proceeds with no visible precipitation. At 2 mg/mL, partial precipitation. At 4 mg/mL, complete precipitation.
Buffer screen at 0.5 mg/mL: Cleavage is clean. Polishing SEC shows a monomer peak with a 5% high-MW shoulder.
Reformulation: Adding 200 mM arginine + 10% glycerol allows cleavage at 2 mg/mL with no visible precipitation.
Scale-up: Final protocol uses on-column cleavage with arginine in the cleavage buffer. Recovery rises from ~5 mg/L of bare kinase to ~15 mg/L.
The protein was always aggregation-prone. MBP-fusion masked the problem at expression. The diagnostic workflow identified that the problem was kinetic (concentration-driven), and on-column cleavage with arginine was the right intervention.
The TEV Cleavage Site Problem: An Underappreciated Variable
Most labs treat TEV cleavage as a black box: add protease, wait, get product. The reality is that the cleavage site itself is a variable that can drive post-cleavage aggregation independent of the bulk surface chemistry of the passenger.
The P1' Residue Constraint
TEV protease recognizes the sequence ENLYFQ↓S, where the ↓ marks the cut. The P1' position (S in the canonical site) becomes the new N-terminal residue of the cleaved passenger. TEV tolerates a range of P1' residues with varying efficiency:
S, G, A, M, C: Efficient cleavage, no significant rate penalty
K, R, H: Reduced cleavage rate, can be problematic at low temperature
N, Q, D, E: Moderate cleavage rate, generally workable
P: Essentially blocks cleavage
F, W, Y, L, I, V: Reduced cleavage with hydrophobic P1', often the worst aggregation outcomes
If your passenger's native N-terminus begins with a hydrophobic residue (a common situation for proteins that begin with an α-helix), you have two bad options: keep the canonical Ser (so the cleaved product has an extra non-native residue) or design a non-canonical site (with a reduced cleavage rate).
The hidden consequence: the non-native Ser at the N-terminus of the cleaved product can itself drive aggregation by disrupting the local folding environment of the first 5–10 residues. Some passengers fold around a particular N-terminal residue identity, and Ser is not it.
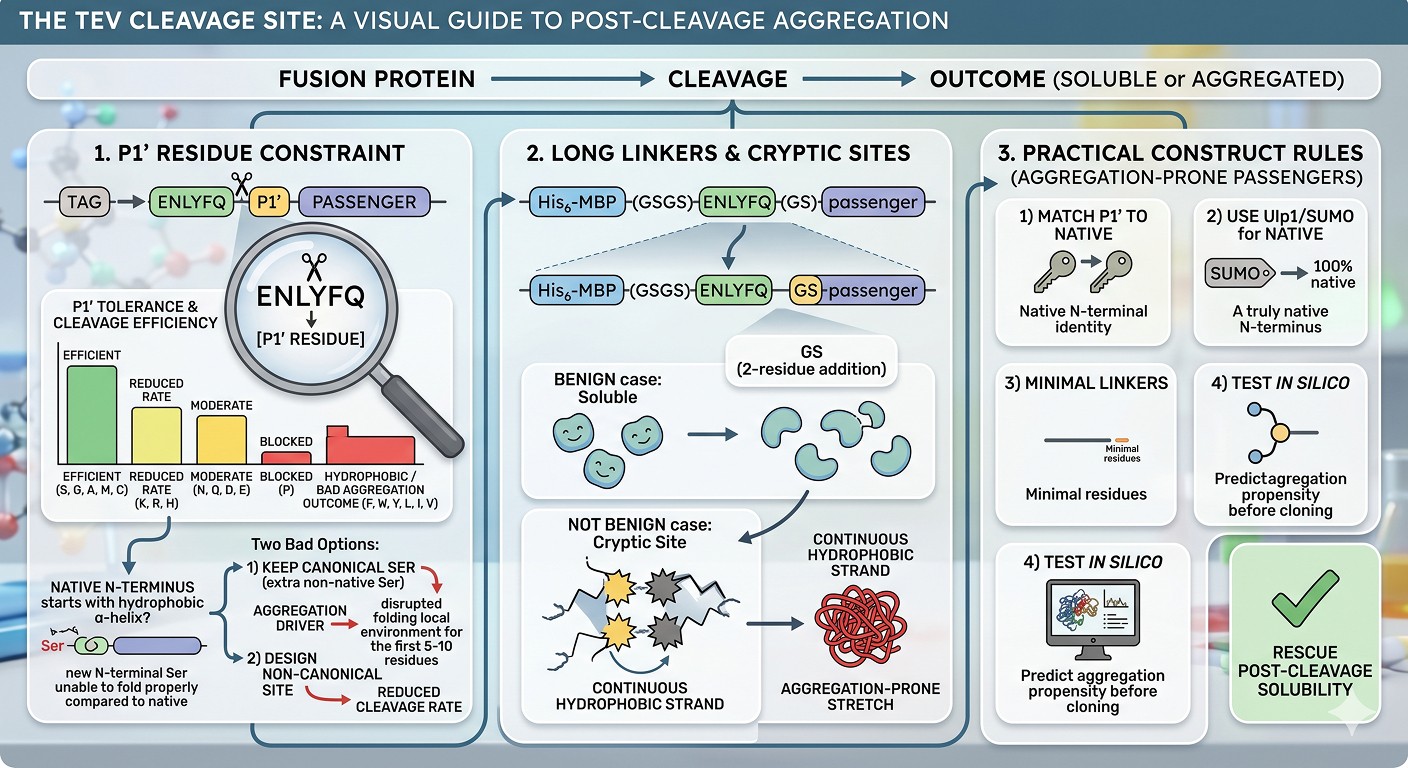
Long Linkers and Cryptic Aggregation Sites
A typical fusion construct includes a short linker between the tag and the TEV site, and sometimes additional residues between the TEV site and the passenger. After cleavage, those "extra" residues remain on the passenger as N-terminal additions. A construct like His₆-MBP-(GSGS)-ENLYFQ-(GS)-passenger leaves GS as a 2-residue N-terminal addition on the passenger.
In most cases this is benign. In some cases it is not. If the residues form an aggregation-prone stretch (for example, if the passenger's native N-terminus begins with hydrophobic residues that pair with the linker residues to form a continuous hydrophobic strand), the construct is destined for trouble.
Practical Construct Rules
For aggregation-prone passengers:
Match P1' to the passenger's native N-terminal residue when possible
Use Ulp1/SUMO if you need a truly native N-terminus
Keep linker residues minimal between the cleavage site and the passenger
Test the post-cleavage construct in silico for aggregation propensity before committing to clone
Why "Soluble Fusion" Can Be Misleading at Lysis
Another version of the paradox: the fusion appears soluble in clarified lysate but is actually present partly as soluble aggregates that survived centrifugation. The standard "soluble vs insoluble" SDS-PAGE on supernatant and pellet is not sensitive to soluble oligomers.
A fusion protein in the 200–500 kDa soluble aggregate range will:
Pellet only partially at 16,000×g
Show as "soluble" on SDS-PAGE because SDS breaks the oligomers apart
Show as a high-MW peak on SEC—but only if the SEC column is run, which is often skipped at the "is it soluble?" stage
Survive IMAC capture and elution
Behave anomalously during cleavage—the protease may cleave the surface-accessible copies first, and the resulting partially-cleaved soluble aggregate becomes seeding-competent
This matters because the standard "MBP fusion is soluble" check misses this failure mode. A protein scientist who runs only the centrifugation-and-SDS-PAGE test will conclude the fusion is fine; the SEC trace would have shown a 30% void-volume peak and predicted the post-cleavage precipitation event.
The recommendation: always run analytical SEC on a fusion before deciding to scale up. If the fusion has a measurable void-volume population, the post-cleavage product will almost certainly precipitate.
Eukaryotic Hosts: When the Paradox Goes Away
The solubility tag paradox is largely a phenomenon of bacterial expression. In insect, mammalian, and yeast hosts, the picture is different:
Eukaryotic chaperone systems (Hsp70, Hsp90, ER chaperones, calnexin/calreticulin) provide ongoing folding assistance that does not depend on a fusion partner
Co-translational folding in eukaryotes is more efficient than in E. coli
ER-targeted proteins benefit from oxidative folding machinery
Glycosylation, when needed, masks hydrophobic surfaces and stabilizes the folded state
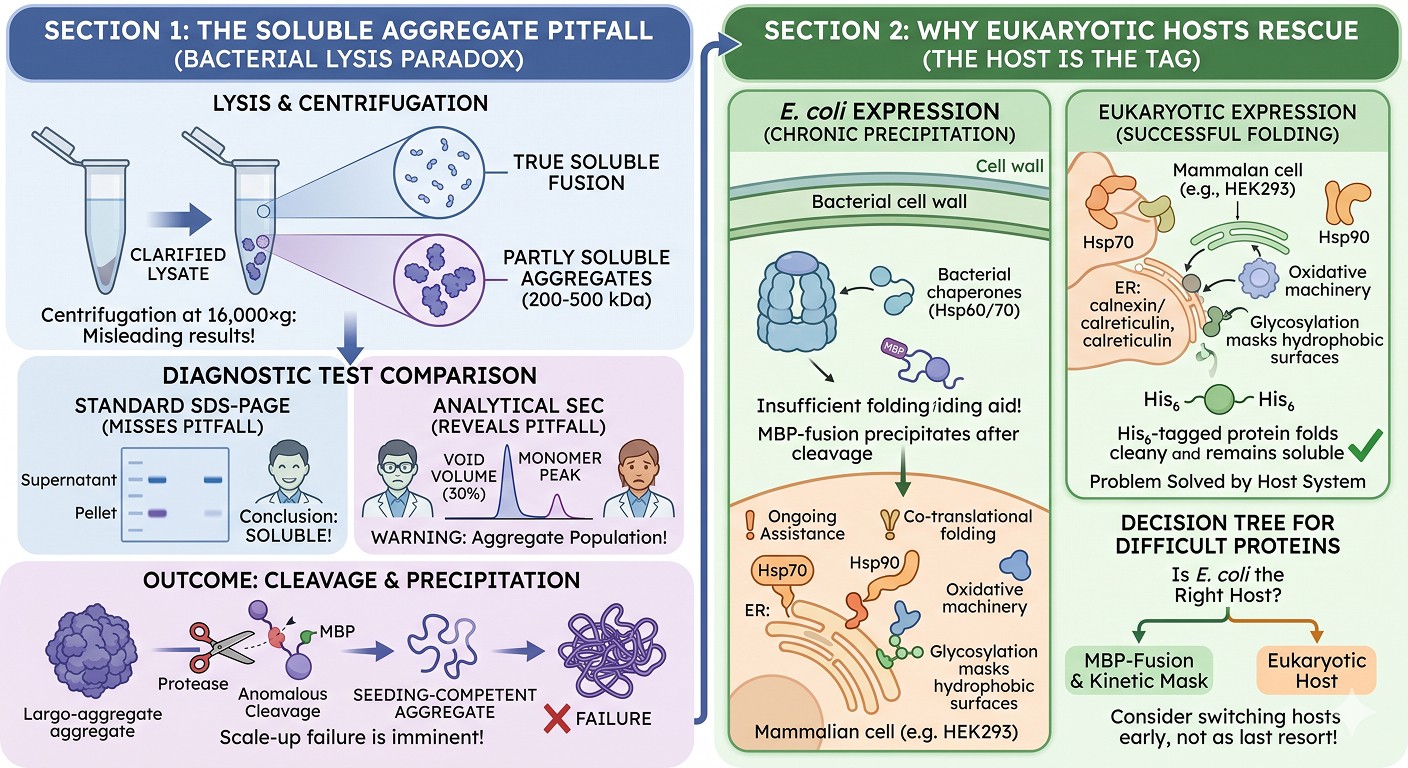
For proteins that require eukaryotic PTMs or chaperone systems, attempting to rescue with bacterial fusion partners is the wrong solution. The diagnostic question is not "which tag should I use" but "is E. coli the right host."
A protein that precipitates after MBP cleavage from E. coli may express cleanly with only a His₆ tag in HEK293 or insect cells, because the underlying problem (a glycosylation requirement, a chaperone-dependent fold, a disulfide-bond requirement) is solved by the host rather than by the tag.
The decision tree for a chronically MBP-cleavage-precipitating protein should include "switch hosts" as a serious option, not a last resort.
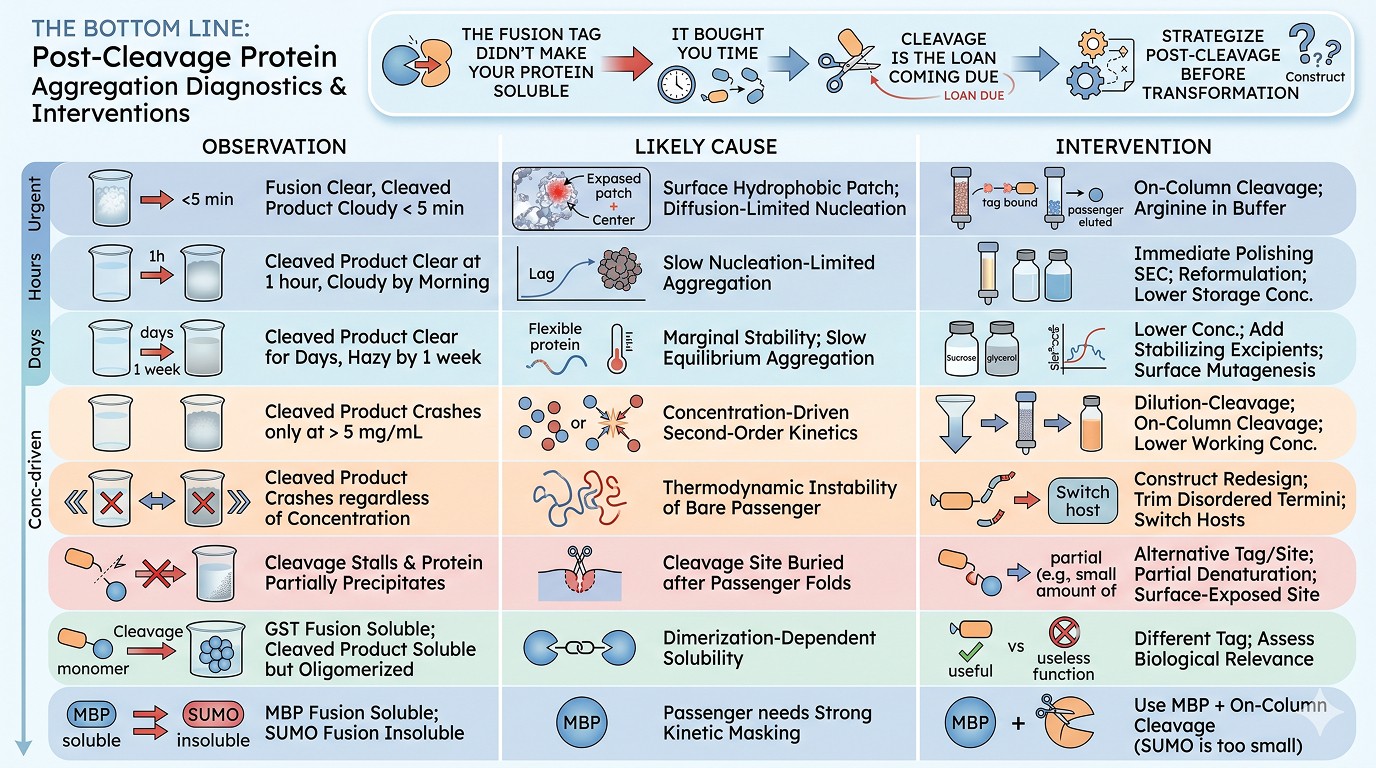
The Bottom Line
Observation | Likely Cause | Intervention |
|---|---|---|
Fusion clear, cleaved product cloudy within 5 minutes | Surface hydrophobic patch; diffusion-limited nucleation | On-column cleavage; arginine in buffer |
Cleaved product clear at 1 hour, cloudy by morning | Slow nucleation-limited aggregation | Immediate polishing SEC; reformulation; lower storage concentration |
Cleaved product clear for days, hazy by 1 week | Marginal stability; slow equilibrium aggregation | Lower concentration; add stabilizing excipients; consider surface mutagenesis |
Cleaved product crashes only at >5 mg/mL | Concentration-driven second-order kinetics | Dilution-cleavage; on-column cleavage; lower target working concentration |
Cleaved product crashes regardless of concentration | Thermodynamic instability of bare passenger | Construct redesign; trim disordered termini; switch hosts |
Cleavage stalls and protein partially precipitates | Cleavage site buried after passenger folds | Try non-native cleavage site; partial denaturation; alternative tag with surface-exposed site |
GST fusion soluble; cleaved product soluble but oligomerized | Dimerization-dependent solubility | Use a different tag; consider whether dimer is biologically relevant |
MBP fusion soluble; SUMO fusion insoluble | Passenger needs strong kinetic masking | Use MBP + on-column cleavage; SUMO is too small for this protein |
The fusion partner did not make your protein soluble. It bought you time. The cleavage event is the moment that loan comes due, and the right strategy is to plan for it before you ever transform the construct.
Designing Constructs That Survive Cleavage With Orbion
Most post-cleavage precipitation is preventable at the construct design stage—if you know what to look for. Orbion's Construct Design module lets you assemble His₆-MBP-TEV-(passenger) and His₆-SUMO-(passenger) constructs in parallel and run each through expression and aggregation prediction before committing to cloning. AstraSUIT identifies whether the passenger's expression-system suitability flags it as a likely "kinetic-mask-dependent" target—proteins predicted to express poorly without fusion partners are precisely the ones most likely to precipitate after cleavage, and the system surfaces this risk before you have invested in fermentation.
AstraUNFOLD flags disordered termini, surface aggregation propensity, and amyloidogenic stretches. A passenger with a high AstraUNFOLD aggregation score at its N-terminus is a candidate for trimming rather than a candidate for "let's hope MBP fixes it." For passengers where redesign is not viable, the Construct Design module also supports tandem-tag and dual-cleavage strategies, so that the rescue interventions described above can be designed in from the start rather than retrofitted at the moment of failure.
References
Kapust RB & Waugh DS. (1999). Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Science, 8(8):1668–1674. PMC22049
Fox JD, Routzahn KM, Bucher MH, Waugh DS. (2003). Maltodextrin-binding proteins from diverse bacteria and archaea are potent solubility enhancers. FEBS Letters, 537(1–3):53–57. DOI
Esposito D & Chatterjee DK. (2006). Enhancement of soluble protein expression through the use of fusion tags. Current Opinion in Biotechnology, 17(4):353–358. PMC2706091
Butt TR, Edavettal SC, Hall JP, Mattern MR. (2005). SUMO fusion technology for difficult-to-express proteins. Protein Expression and Purification, 43(1):1–9. DOI
Costa S, Almeida A, Castro A, Domingues L. (2014). Fusion tags for protein solubility, purification and immunogenicity in Escherichia coli: the novel Fh8 system. Frontiers in Microbiology, 5:63. PMC3933005
LaVallie ER, et al. (1993). A thioredoxin gene fusion expression system that circumvents inclusion body formation in the E. coli cytoplasm. Nature Biotechnology, 11:187–193. DOI
Arakawa T, Ejima D, Tsumoto K, Obeyama N, Tanaka Y, Kita Y, Timasheff SN. (2007). Suppression of protein interactions by arginine: a proposed mechanism of the arginine effects. Biophysical Chemistry, 127(1–2):1–8. DOI
Waugh DS. (2011). An overview of enzymatic reagents for the removal of affinity tags. Protein Expression and Purification, 80(2):283–293. PMC3188689
Book a 20-Minute Demo
Sign up free for unlimited Overview runs — summary, sequence-based analysis, homology search. For the full Characterization — PTMs, binding sites, stability variants, construct design — book a demo and we'll run your target live.
