Blog
Orbion Team
What AlphaFold3 Changes for Binding and Complex Prediction — and What It Doesn't

You fed your kinase and a lead fragment into the AlphaFold3 server, and 90 seconds later you had an all-atom protein–ligand complex with the ligand sitting in the pocket. No docking grid, no receptor preparation, no ensemble of scoring functions. For a structural biologist or medicinal chemist who spent the last decade wrestling with docking pipelines, this feels like the rules just changed.
They did — but not in every direction you might hope. AlphaFold3 co-folds proteins, ligands, ions, nucleic acids, and modified residues in a single pass, and it beats classical docking on the PoseBusters benchmark by a wide margin. It also gives you no binding affinity, hallucinates poses for genuinely novel ligands, and occasionally hands you a structure with inverted chirality. This piece is about exactly where the line falls between the two.
Key Takeaways
AlphaFold3 co-folds the whole assembly at once: proteins, small molecules, ions, nucleic acids, and modified residues share one diffusion-based prediction, replacing the separate docking step entirely for many cases.
It beats classical docking on pose accuracy: AlphaFold3 produces significantly more pocket-aligned ligand RMSD < 2 Å poses than AutoDock Vina or RoseTTAFold All-Atom on the PoseBusters set (Fisher's exact test, P = 2.27 × 10⁻¹³ vs Vina).
It does not output binding affinity: AlphaFold3 predicts a pose and a confidence, not a Kd, IC50, or ΔG. Confidence is geometric agreement, not thermodynamics.
Novel ligands and disordered regions are where it breaks: the diffusion model is generative, so it can hallucinate plausible-looking but wrong order, and it still violates ligand chirality in about 4.4% of benchmark cases.
Confidence metrics carry real caveats: high ipTM on a full-length input does not prove a true interface — AlphaFold can report misleadingly high ipTM for non-interacting protein pairs, particularly when disorder is present in both chains [9].
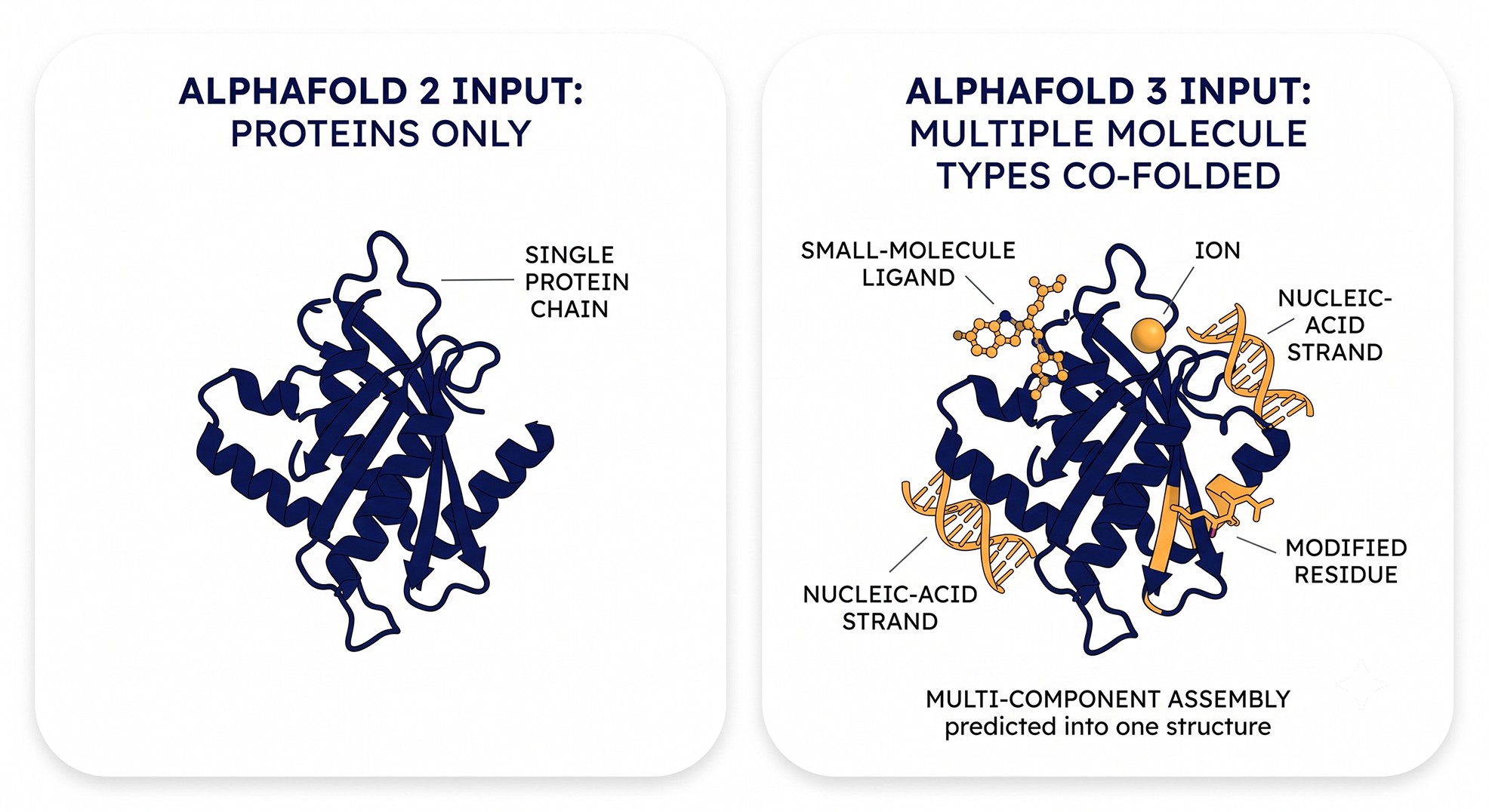
What AlphaFold3 Actually Adds Over AlphaFold2 and Multimer
To see what's new, it helps to be precise about what AlphaFold2 and AlphaFold-Multimer could and couldn't do.
AlphaFold2 (2021) predicts single-chain protein structures from sequence with atomic accuracy [1]. AlphaFold-Multimer (2022) extends that to protein–protein complexes of known stoichiometry, predicting interfaces between polypeptide chains [2]. Both are protein-only. Neither models a small-molecule ligand, a metal ion, a nucleotide, or a phosphorylated serine as a first-class entity. If you wanted a ligand in the pocket, you docked it afterward or transplanted it from a homolog.
AlphaFold3 (2024) changes the input space and the architecture at the same time [3].
Joint all-atom co-folding
The headline change: AlphaFold3 predicts the joint structure of a complex that can include proteins, nucleic acids (DNA and RNA), small-molecule ligands, ions, and chemically modified residues — all in one prediction [3]. You are no longer predicting a protein and then placing a ligand. You are predicting the protein and the ligand and the ions together, with each influencing the other's coordinates.
This matters for medicinal chemistry because the receptor is not a rigid grid you dock into. Side chains in the pocket can rearrange around the ligand during co-folding, which is closer to what happens physically than docking into a fixed structure.
A diffusion architecture that works on raw atoms
AlphaFold2's structure module predicted residue frames and torsions — a representation built around the protein backbone. That representation has no natural way to describe an arbitrary small molecule.
AlphaFold3 replaces it with a diffusion module that operates directly on raw atom coordinates, without backbone rotational frames [3]. Diffusion means the model starts from noise and iteratively denoises toward a structure, the same generative principle behind image-generation models. Because it works at the level of atoms rather than residue frames, it can represent any chemical component — a benzene ring, a zinc ion, a methylated lysine — using the same machinery it uses for protein backbone atoms.
This architectural choice is what makes the broadened input space possible. It also introduces the model's most important new failure mode, which we'll get to.
Better protein–ligand poses than docking
On the PoseBusters benchmark — 428 recent protein–ligand complexes from the PDB, designed to test both geometric accuracy and physical validity [4] — AlphaFold3 produces a substantially higher fraction of correct poses (pocket-aligned ligand RMSD < 2 Å) than both a classical docking tool (AutoDock Vina) and a deep-learning all-atom method (RoseTTAFold All-Atom). The paper reports Fisher's exact test P = 2.27 × 10⁻¹³ versus Vina and P = 4.45 × 10⁻²⁵ versus RoseTTAFold All-Atom [3].
That is a real, measurable improvement on the specific task of placing a known ligand in a known pocket. Note the framing: known ligand, structurally represented in training-adjacent PDB space.
Better antibody–antigen interfaces
AlphaFold-Multimer historically struggled with antibody–antigen complexes — the CDR loops that define specificity are hypervariable and poorly constrained by evolutionary signal. AlphaFold3 reports substantially higher antibody–antigen accuracy than AlphaFold-Multimer v2.3 (paired Wilcoxon signed-rank test, P = 6.5 × 10⁻⁵), with the caveat that this gain depends on generating and ranking many model seeds [3].
The core shift: AlphaFold2/Multimer answered "what does this protein (or protein complex) look like?" AlphaFold3 answers "what does this assembly look like?" — where the assembly can include the drug-like molecule you actually care about.
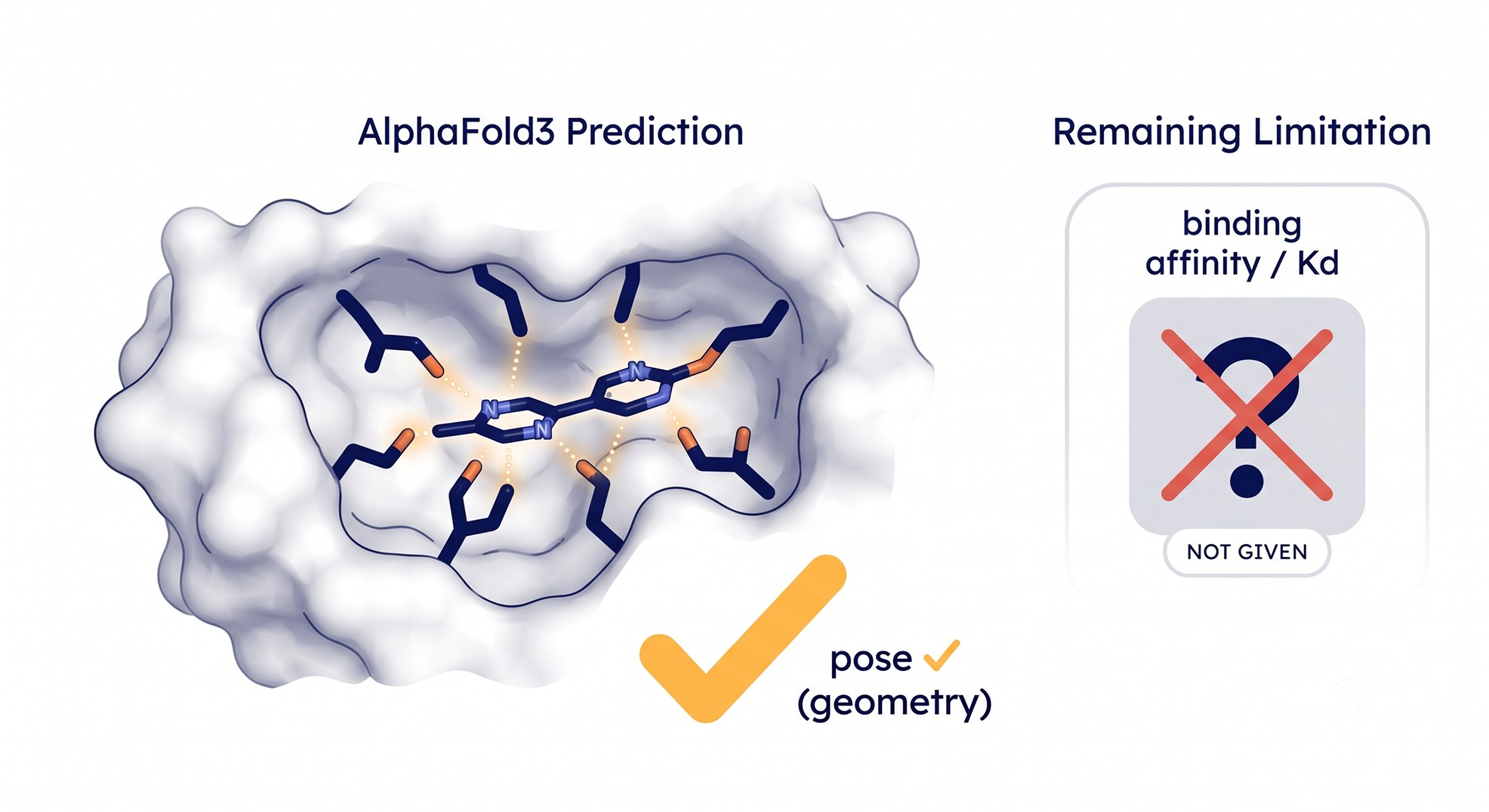
What AlphaFold3 Still Doesn't Do
Here is where careful reading of the 2024 Nature paper saves you from expensive mistakes.
It does not predict binding affinity
This is the single most common misconception, and it costs the most. AlphaFold3 gives you a pose and a confidence score. It does not give you a Kd, an IC50, a ΔG, or any thermodynamic quantity [3].
A confident pose tells you the model thinks the ligand sits there. It says nothing about how tightly it binds, or whether a chemically similar analog would bind tighter or weaker. You cannot rank a congeneric series by potency using AlphaFold3 confidence. The model will happily co-fold a non-binder into a pocket and report a confident-looking pose, because "does this molecule fit geometrically" and "does this molecule bind" are different questions.
Recent work bears this out in both directions. For covalent binders, AF3 co-folding plus its predicted confidence metric can classify true binders against property-matched decoys with near-optimal accuracy on a kinase benchmark — outperforming state-of-the-art covalent docking — and even surfaced a novel BTK binder in a prospective screen [5]. But that is a binary classification task on covalent chemistry, not a general affinity readout, and it does not generalize to ranking a non-covalent SAR series by potency.
What it can do: tell you a plausible binding mode for a known or near-known ligand.
What it cannot do: tell you affinity, rank a potency series, or substitute for an assay.
It hallucinates structure for novel ligands and disordered regions
Because the diffusion module is generative, it always produces a structure — even when the right answer is "disordered" or "doesn't bind." AlphaFold2 was non-generative and tended to render disordered regions as recognizable extended ribbons. AlphaFold3 can instead invent confident-looking spurious order, sometimes folding a genuinely disordered stretch into a plausible helix [6].
The DeepMind team anticipated this and added cross-distillation — enriching training data with AlphaFold-Multimer predictions so the model learns to represent unstructured regions as extended loops rather than fabricated folds [3]. It mitigates the problem; it does not eliminate it. Hallucinated regions usually carry very low confidence (pLDDT well below 50), so the signal is there — but only if you read it [6].
For genuinely novel chemotypes — ligands with no close analog in the PDB — pose reliability drops. The model is strongest where the training distribution is dense and weakest exactly where a discovery chemist most wants help: novel scaffolds against under-characterized pockets.
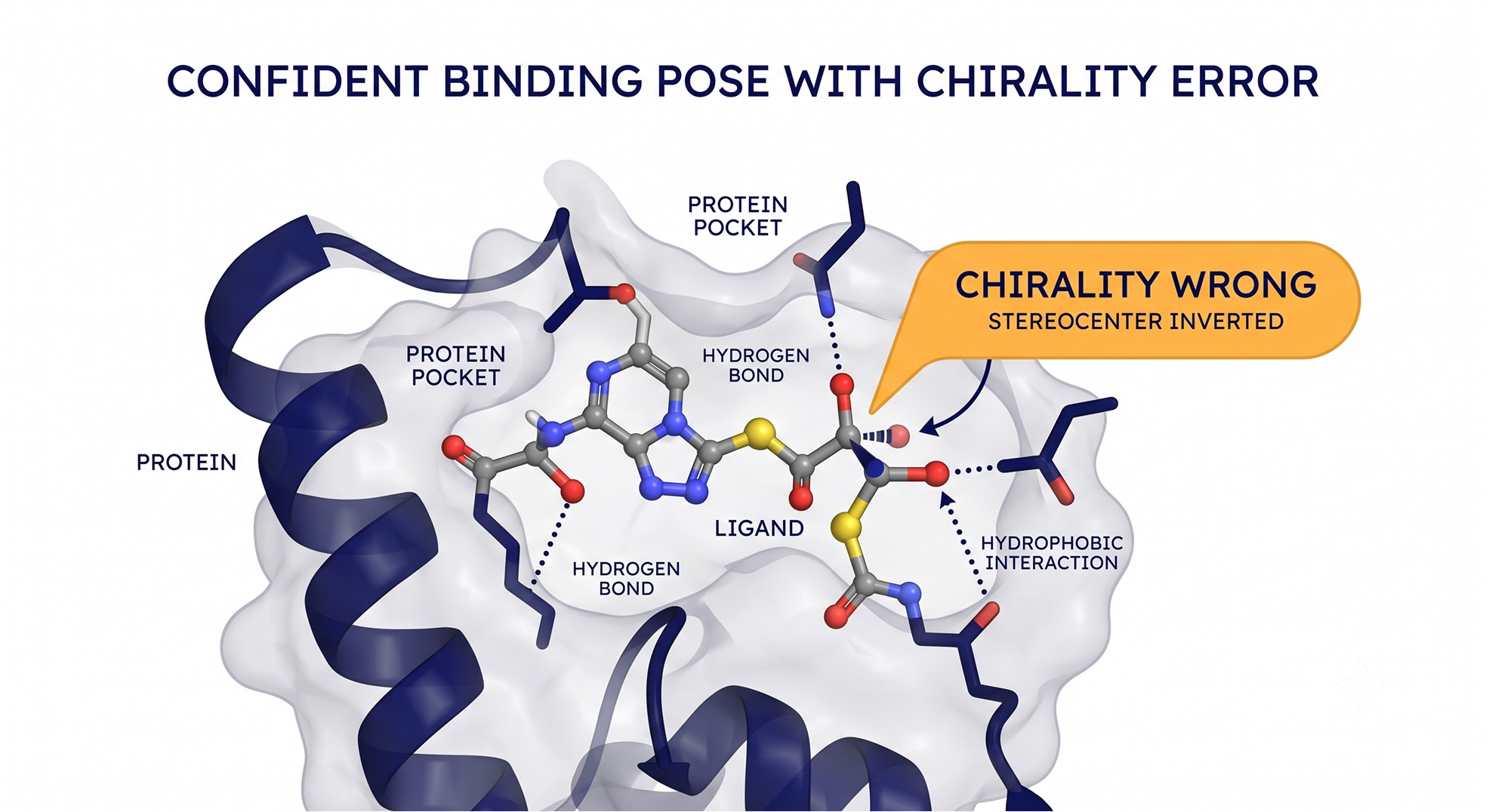
It violates stereochemistry and clashes atoms
A diffusion model denoising atom coordinates has no hard-wired guarantee of chemical validity. The result: AlphaFold3 outputs sometimes contain chirality violations — even when given a reference structure with correct chirality as input. The AlphaFold3 paper itself reports a chirality violation rate of 4.4% on its PoseBusters benchmark, despite penalizing chirality mismatches during ranking [3]; subsequent work confirms and characterizes these stereochemical errors in detail [7]. Inverted stereocenters, distorted bond lengths and angles, and occasional atomic clashes all appear.
The ranking formula includes terms that penalize clash-heavy predictions [3], which helps the top-ranked model but does not make the underlying outputs chemically clean. For a medicinal chemist, an inverted stereocenter is not a rounding error — it can be the difference between an agonist and an inactive enantiomer. Always run a cheminformatics validity check (PoseBusters-style: chirality, bond geometry, intramolecular clashes, ligand–protein overlap) before you trust a pose [4].
It predicts one static structure, not dynamics
AlphaFold3 predicts a single static assembly. It does not give you a conformational ensemble, an induced-fit trajectory, or the population of states a flexible protein samples in solution [6]. For cryptic pockets, allosteric transitions, or fragment-to-lead campaigns where binding reshapes the receptor, one static snapshot is a starting hypothesis, not the full picture.
Access and licensing constrain how you use it
The AlphaFold3 server offers free non-commercial access, and the inference code was released under terms that restrict commercial use; for a long initial period the server did not even accept arbitrary user-supplied ligands. If you are in a pharma or biotech setting, read the licensing carefully — it shapes whether AlphaFold3 can sit inside a production discovery pipeline at all, and it is the reason many teams run AlphaFold2/Multimer-class models in-house for routine work.

Reading AlphaFold3 Confidence Without Fooling Yourself
AlphaFold3 reports four confidence signals [3]:
Metric | What it measures | What it does not mean |
|---|---|---|
pLDDT | Per-atom local confidence (0–100) | Not global correctness; high pLDDT on a wrong fold is possible |
PAE | Predicted aligned error between residue/atom pairs | Not a contact map; large PAE = uncertain relative positioning |
pTM | Predicted global fold accuracy (TM-score) | Says little about a specific interface |
ipTM | Predicted interface accuracy | Not binding affinity, and not proof an interface is real |
The ipTM caveat is the one that burns people. ipTM can stay misleadingly high for protein pairs that do not actually interact — especially when both chains carry disordered or non-interacting accessory regions — so the score does not, on its own, establish that a predicted interface is real or that the proteins bind [9]. High ipTM is necessary, not sufficient: always corroborate a predicted interface with orthogonal evidence (co-evolutionary signal, pull-down, SPR, or a known biological role).
Treat confidence as a triage signal that flags predictions worth experimental follow-up — never as a substitute for the experiment.

Case Study: A Co-Folded Pose That Looked Perfect and Was Wrong
Problem: A medicinal chemistry team co-folded a serine hydrolase with a novel macrocyclic inhibitor on the AlphaFold3 server. The output was striking — the macrocycle nestled in the catalytic pocket, pLDDT in the pocket residues above 85, and a clean-looking complex they were ready to use to justify the next synthesis round.
Analysis: Before committing, they ran two checks. First, a PoseBusters-style validity scan flagged a chirality violation at one stereocenter of the macrocycle — the model had inverted it relative to the synthesized compound. Second, they noted that the ligand was a genuinely novel chemotype with no close analog in the PDB, putting it squarely in the model's weak regime. The confident-looking pocket pLDDT was reporting on the protein, not validating the ligand placement.
Solution: They re-derived the binding hypothesis with an orthogonal method — classical docking with the correct stereochemistry enforced, cross-checked against a homolog co-crystal structure, and an AlphaFill-style transplant of the cofactor from a related experimental structure to anchor the active site. They also commissioned a low-resolution competition assay before scaling synthesis.
Outcome: The corrected analysis placed the macrocycle in a shifted sub-pocket, ~4 Å from the AlphaFold3 pose, and the assay confirmed weak binding rather than the potent interaction the confident pose had implied. The team avoided three synthesis cycles chasing a geometry artifact. The lesson: a confident AlphaFold3 protein is not a validated ligand pose, and novel chemotypes demand orthogonal confirmation.
A Decision Tree for Using AlphaFold3 on Binding Problems
AlphaFold2/Multimer vs AlphaFold3: What You Gain and What You Pay
Dimension | AlphaFold2 / Multimer | AlphaFold3 |
|---|---|---|
Input space | Proteins only (single chain / complexes) | Proteins + ligands + ions + nucleic acids + modified residues |
Architecture | Residue-frame structure module | Diffusion module on raw atom coordinates |
Protein–ligand poses | Not supported (dock separately) | Beats Vina & RoseTTAFold-AA on PoseBusters [3] |
Antibody–antigen | Weak | Improved (needs many seeds) [3] |
Binding affinity | None | Still none |
Hallucination risk | Lower (non-generative; ribbon-like disorder) | Higher (generative; can fabricate order) [6] |
Chemical validity | N/A for ligands | Chirality/clash violations occur (4.4% chirality on PoseBusters) [3] |
Output | One static structure | One static structure |
In-house deployment | Mature, widely self-hosted | Server access + licensing constraints |
The honest read: AlphaFold3 expands what you can model and improves pose accuracy, but it does not change the two hardest problems in drug discovery — predicting affinity and predicting dynamics. Those remain assay-and-simulation territory.
Practical Checklist Before You Trust an AlphaFold3 Complex
Is the ligand a known chemotype with a close PDB analog? (If not, downgrade trust.)
Did you run a chirality / bond-geometry / clash validity check on the ligand?
Did you read pLDDT on the pocket and ligand, not just the global protein?
For interfaces: did you generate many seeds and read ipTM with skepticism?
Are you treating confidence as a triage flag, not as affinity?
Did you cross-check against any homolog co-crystal structure?
Do you have an orthogonal plan (assay, docking, MD) before committing synthesis or resources?
Does your licensing context permit the intended (commercial?) use?
Bottom Line
AlphaFold3 made co-folding proteins with ligands, ions, and nucleic acids routine, and it genuinely beats docking on pose accuracy — but it predicts geometry, not affinity, and it hallucinates exactly where you most need help: novel ligands, disordered regions, and dynamics. Use it as a fast, powerful hypothesis generator. Validate before you bet on it.
How Orbion Helps
The field is clearly moving toward AF3-class co-folding, and that direction is the right one for binding and complex problems. Today, Orbion gives you binding and interface insight built on AlphaFold2 and AlphaFold-Multimer — with the confidence-reading discipline this article argues for baked in, so you're not handed a pretty structure and left to guess what it means.
Orbion's Characterization module predicts structure with full pLDDT per-residue and PAE matrices, and its PAE Insight Engine turns those confidence metrics into interpretable signals — domain boundaries, hinge regions, and conformational heterogeneity cues — rather than leaving you to eyeball a PAE plot. For ligands and cofactors, AlphaFill transplants small molecules and ions from homologous experimental structures into your predicted model [8], giving you a physically grounded placement anchored in real crystallographic data — a complementary, evidence-based answer to the co-folding question. AstraBIND predicts ligand-binding sites and likely ligand types directly from sequence and structural context, aggregating multiple models so you know where to look before you commit to a pose. For complexes, Orbion runs AlphaFold-Multimer with interface analysis (interface area, contact residues, buried surface) and inter-chain PAE — the same confidence signals AlphaFold3 reports, read with the same caution.
Relevant Orbion features:
Characterization with pLDDT + PAE: structure prediction with per-residue and inter-residue confidence you can actually interpret.
PAE Insight Engine: converts AlphaFold2 confidence into domain boundaries, hinges, and dynamics cues — the structural-uncertainty signal a single static pose hides.
AlphaFill enrichment: ligand and cofactor transplant from homologous PDB structures, grounding binding hypotheses in experimental data.
AstraBIND: sequence-and-structure ligand binding-site prediction to localize pockets before pose generation.
AlphaFold-Multimer interface analysis: contact residues, buried surface, and inter-chain PAE for protein–protein and complex work.
Want to localize a binding site and read your structure's confidence honestly before you trust any pose? Paste a sequence into Orbion and start with Characterization.
References
Jumper J, Evans R, Pritzel A, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873):583–589. Link
Evans R, O'Neill M, Pritzel A, et al. (2022). Protein complex prediction with AlphaFold-Multimer. bioRxiv (preprint). Link
Abramson J, Adler J, Dunger J, et al. (2024). Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature, 630(8016):493–500. Link
Buttenschoen M, Morris GM, Deane CM. (2024). PoseBusters: AI-based docking methods fail to generate physically valid poses or generalise to novel sequences. Chemical Science, 15(9):3130–3139. Link
Shamir Y, Gabizon R, Rogel A, Lin DY, Andreotti AH, London N. (2025). Discovery of covalent ligands with AlphaFold3. Journal of the American Chemical Society. Link
EMBL-EBI. (2024). What AlphaFold 3 struggles with — AlphaFold 3 and AlphaFold Server (online training). EMBL-EBI Training. Link
Ishitani R, Moriwaki Y. (2025). Improving Stereochemical Limitations in Protein–Ligand Complex Structure Prediction. ACS Omega. Link
Hekkelman ML, de Vries I, Joosten RP, Perrakis A. (2023). AlphaFill: enriching AlphaFold models with ligands and cofactors. Nature Methods, 20(2):205–213. Link
Dunbrack RL Jr. (2025). Rēs ipSAE loquuntur: what's wrong with AlphaFold's ipTM score and how to fix it. bioRxiv (preprint). Link
Book a 20-Minute Demo
Sign up free for unlimited Overview runs — summary, sequence-based analysis, homology search. For the full Characterization — PTMs, binding sites, stability variants, construct design — book a demo and we'll run your target live.
