Research
Aniruddh Goteti
Can the current tools predict protein epistasis?

We thought protein epistasis was everywhere. Turns out, much of it was an illusion.
If you mutate a protein twice, can the second mutation behave differently because of the first?
That question sits at the heart of epistasis. It matters for protein engineering, evolution, disease biology, and almost any attempt to predict what happens when biology changes in more than one place at once. If we want to design better enzymes, understand resistance mutations, or build models that can actually reason about proteins, we need to understand epistasis.
So we went big.
We assembled what is, to our knowledge, the largest open protein epistasis dataset to date: 6.9 million variant measurements across 256 proteins, collected from 15 public data sources. From that, we computed epistasis for 962,375 double mutants across 176 proteins, and then asked a simple but important question:
Can ESM-2 predict protein epistasis in a zero-shot setting?
At first glance, the answer looked like yes.
But that answer was wrong.
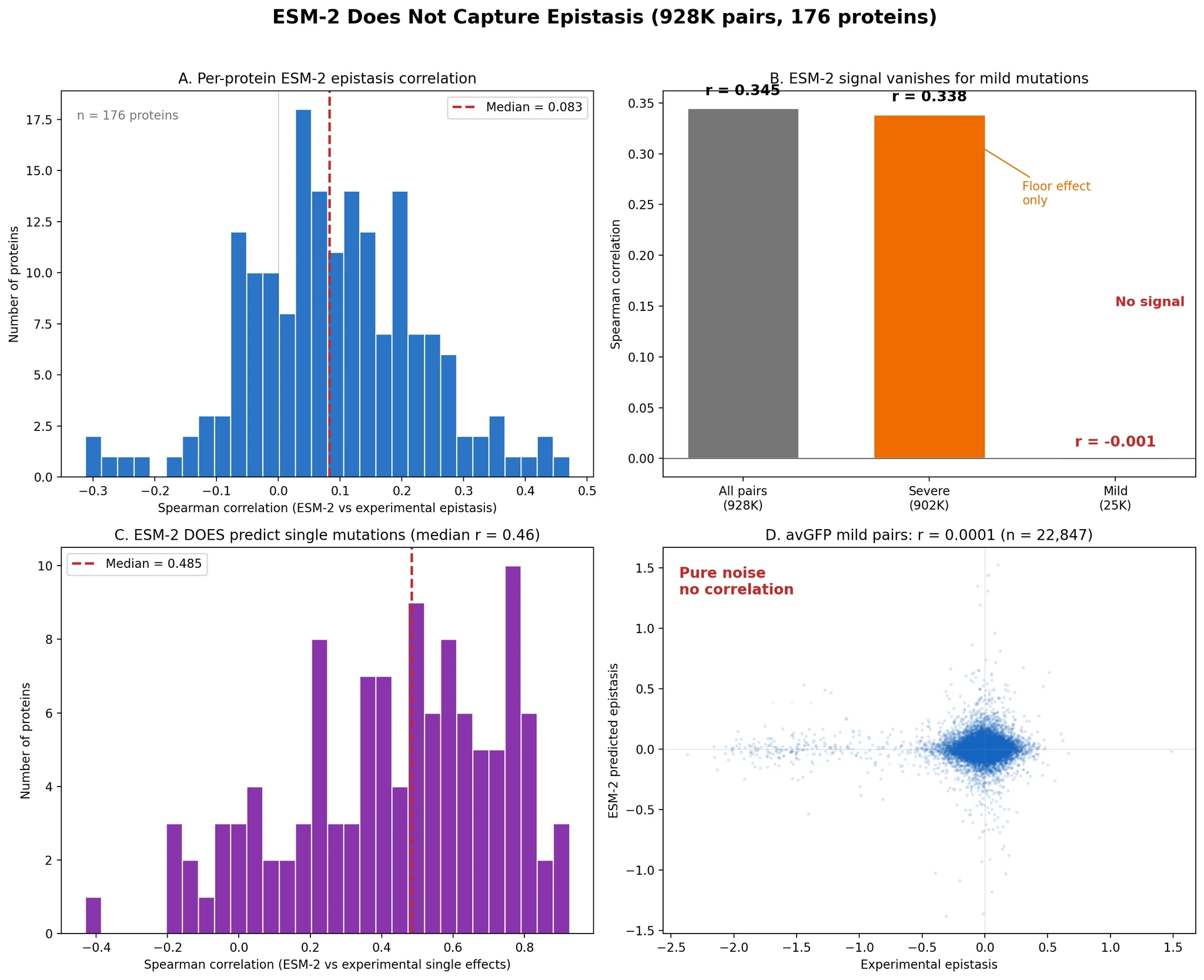
Figure 1. Overview of the dataset and the overall epistasis landscape across 6.9M variant measurements.
First, what is epistasis?
Let’s make this intuitive.
Imagine a protein is like a machine with many tiny parts. A mutation changes one part. Sometimes that change helps. Sometimes it hurts. Sometimes it barely matters.
Now imagine making two mutations at once.
If the effect of the double mutation is just the sum of the two single mutations, that is additive behavior.
But if the double mutation behaves differently than expected, that is epistasis.
So epistasis is biology’s way of saying:
“These parts are interacting. You cannot understand the whole system by looking at each mutation alone.”
That is why epistasis matters so much. It is where simple prediction starts to fail.
How common does epistasis look?
In the raw data, epistasis looked extremely common. The per protein median was very high, and sign epistasis also appeared widespread.
That sounds dramatic. It makes proteins seem full of hidden, tangled interactions.
But as we will see, a lot of that apparent signal is inflated by measurement artifacts.
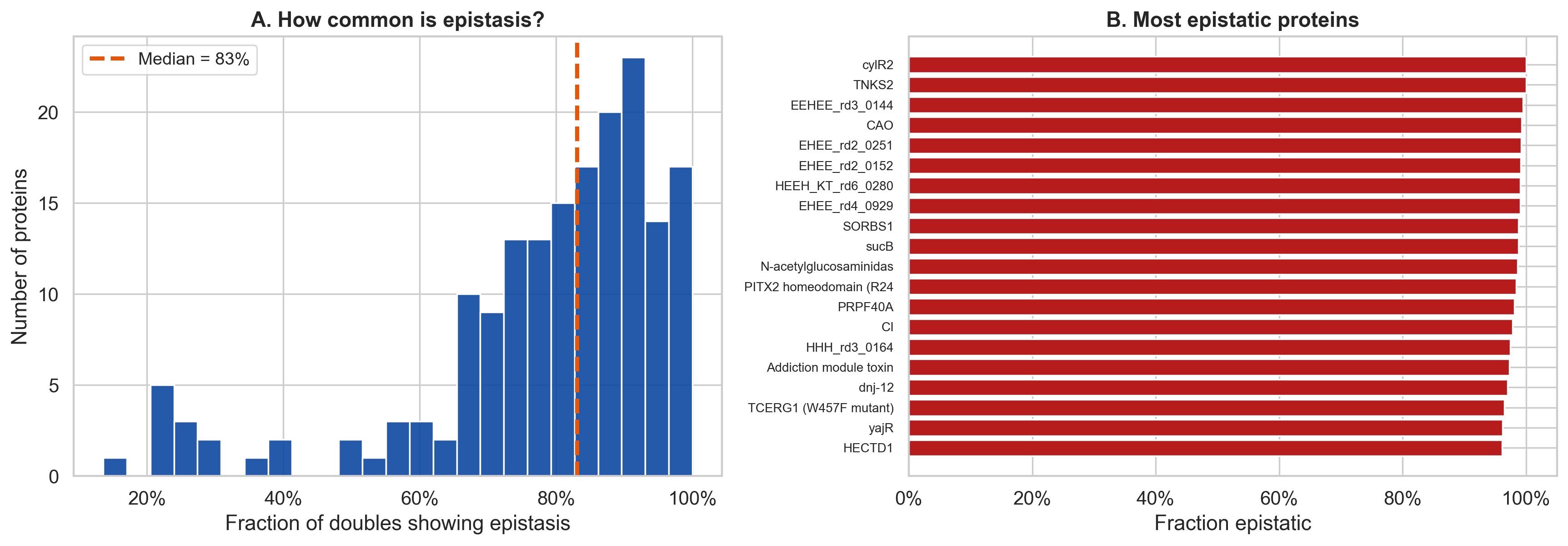
Figure 2. In raw measurements, epistasis appears extremely common across proteins, with a high median fraction of double mutants showing non additive behavior.
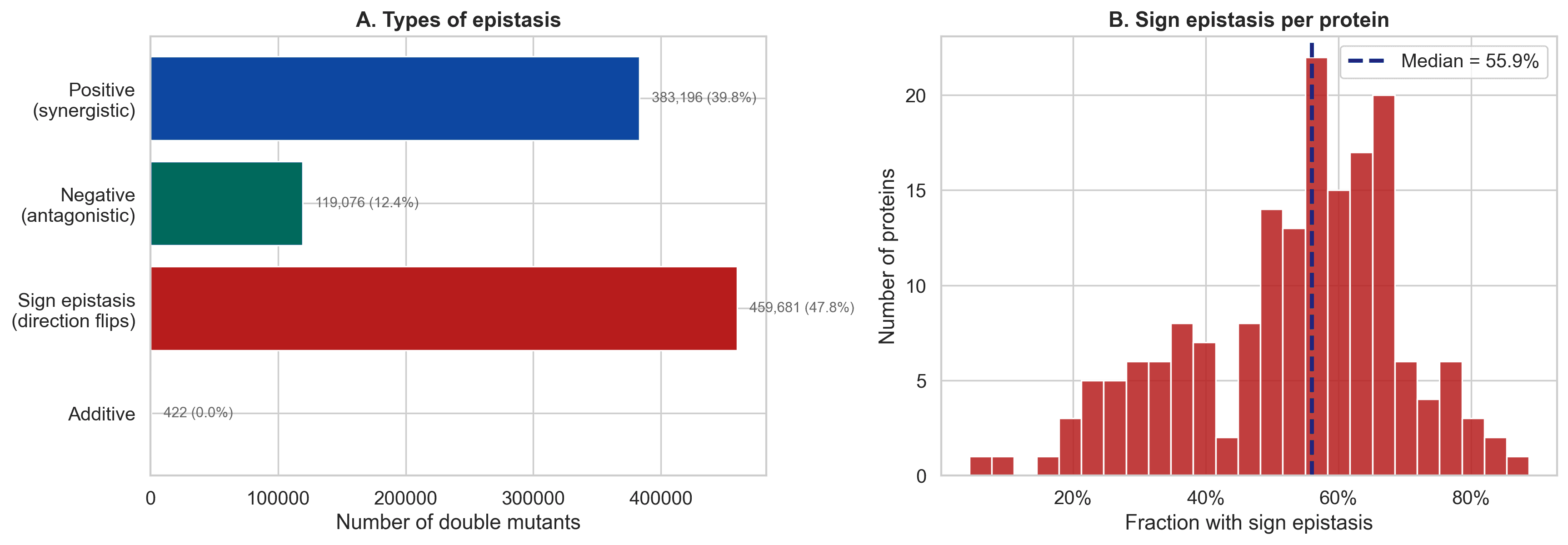
Figure 3. Sign epistasis is especially important because it means the combined outcome can flip direction relative to what additive reasoning would suggest.
The big surprise: much of reported epistasis is not real interaction
Across the raw data, epistasis looked extremely common. The apparent rate was about 59%.
But once we looked more carefully, we found something important:
A lot of that signal was coming from a very boring source.
It was a measurement artifact called the floor effect.
The floor effect, explained simply
Here is the basic idea.
If a single mutation already destroys a protein, the protein is already near the bottom. It cannot get much worse.
So when you add a second harmful mutation, the additive model may predict something even lower, but reality cannot go below the floor. The protein is already basically dead.
That makes the double mutant look better than expected.
Not because the two mutations are doing something clever together.
Just because you already hit bottom.
This is the core reason raw epistasis estimates can be badly inflated.
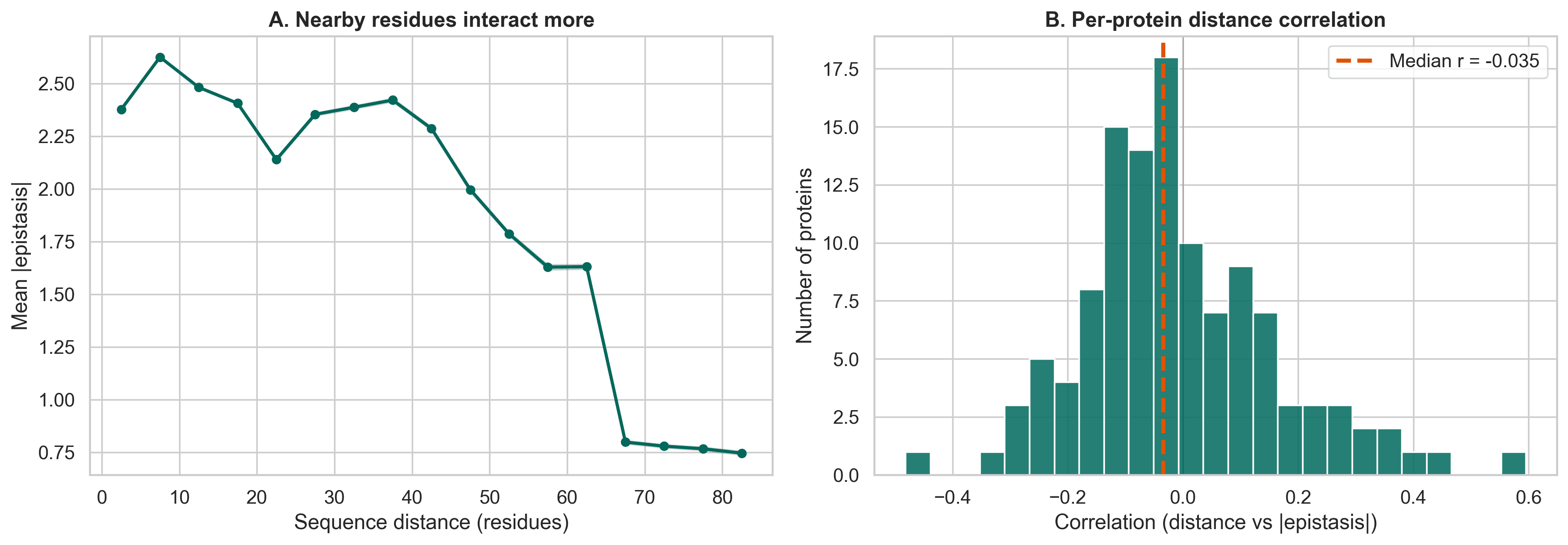
Figure 4. Severe mutations generate artificial positive epistasis. Once those cases are removed, the apparent epistasis rate drops from 59.4% to 21.9%.
What happens when you remove the artifact?
This is where the story gets much more interesting.
When we restrict the analysis to mild single mutations, where floor effects cannot dominate, the apparent epistasis rate drops from 59.4% to 21.9%. Positive epistasis also collapses from an overwhelming majority to something much closer to balanced.
That means proteins are not behaving like chaotic magic.
But they are also not simple.
The cleaner picture is:
real epistasis is less common than many raw analyses suggest
the epistasis that remains is more biologically meaningful
and it becomes much easier to interpret structurally
That is a much better scientific result than just saying that everything interacts with everything.
A field-wide problem, not just one dataset
To put this in context, published epistasis rates across protein studies vary widely. A big part of that variation comes from whether mutation severity and floor effects are properly controlled.
Our contribution was to show this systematically at scale, across 176 proteins and 962,375 double mutants, while also testing ESM-2 zero-shot epistasis prediction on 928,506 mutation pairs.
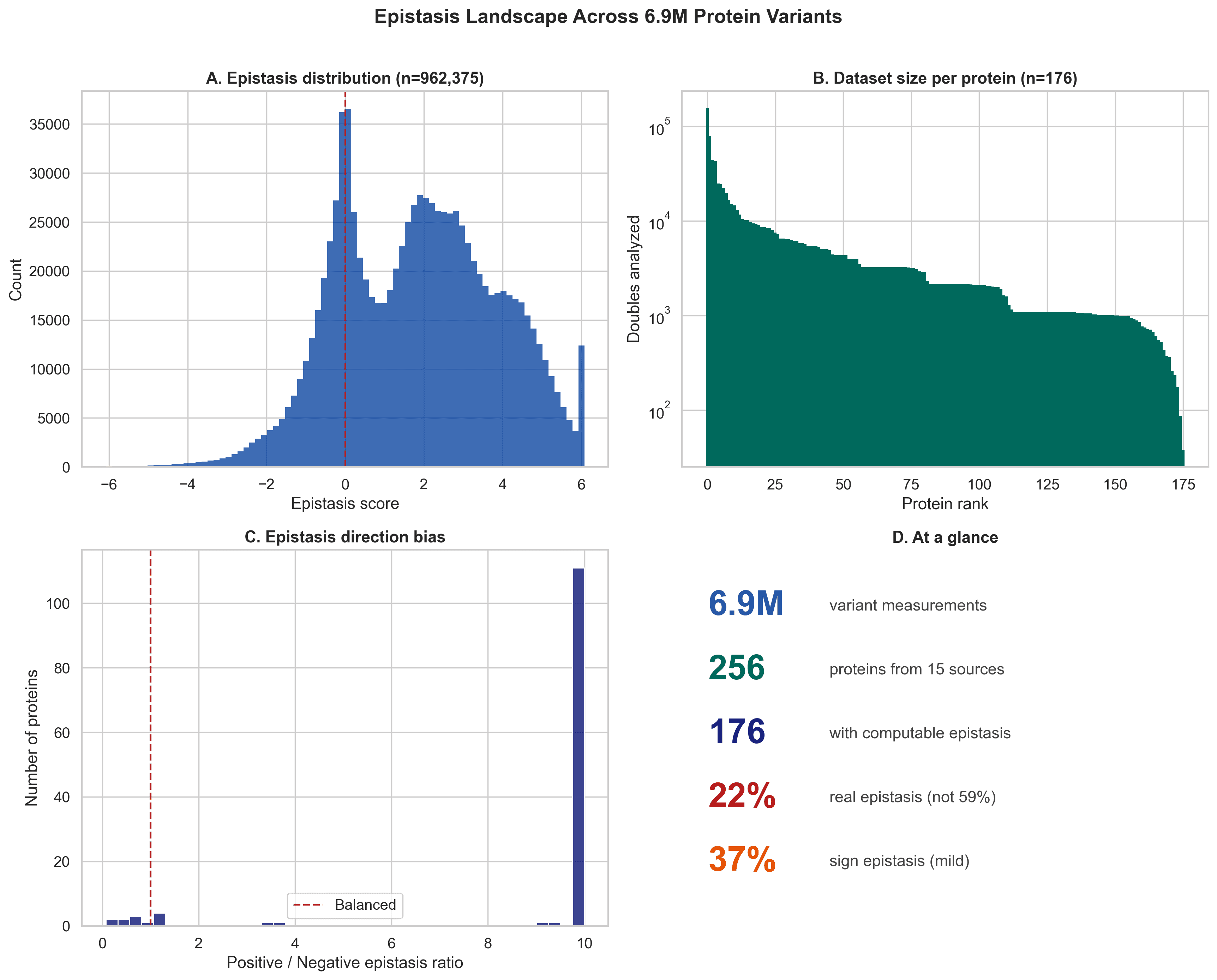
Figure 5. Compared with prior work, this study covers many more proteins and shows that floor corrected epistasis rates are far lower than many raw reports suggest.
The strongest result: ESM-2 does not actually predict epistasis
This was the central test.
We scored 928,506 mutation pairs with ESM-2 650M using a zero-shot setup. Across all pairs, the model showed a seemingly impressive correlation. If you stop there, it looks like ESM-2 captures epistasis pretty well.
But once we remove the floor effect cases and keep only mild mutation pairs, the signal disappears.
That is not “weak but promising.”
That is no signal.
So what was the model picking up in the full dataset?
Mostly the floor effect.
It was learning when proteins were already broken, not when two mutations were truly interacting in a biologically meaningful way.
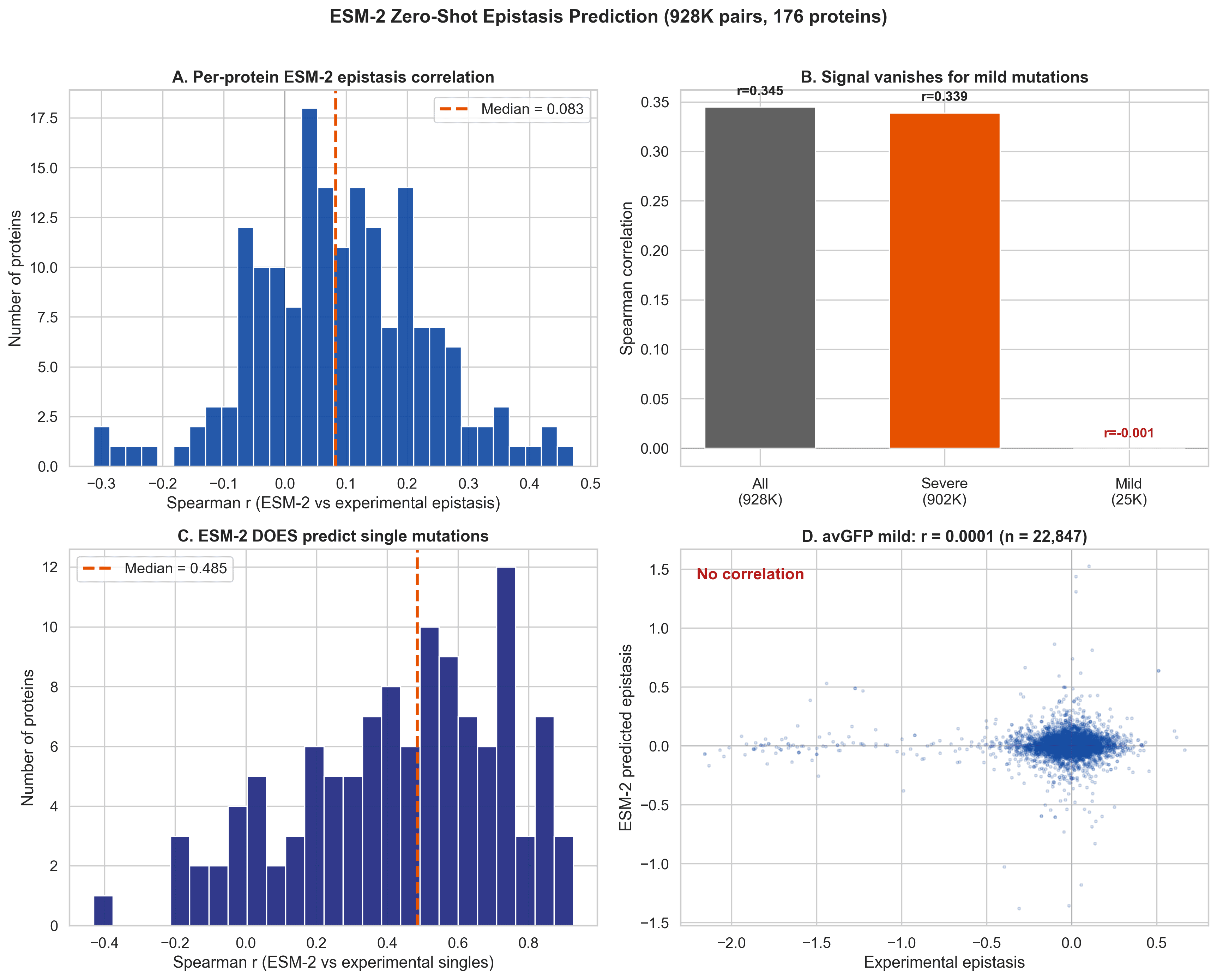
Figure 6. The apparent overall signal vanishes for mild mutation pairs. Meanwhile, ESM-2 still predicts single mutation effects reasonably well.
To be fair: ESM-2 is not useless
This negative result is important, but it is also important not to overstate it.
ESM-2 does predict single mutation effects reasonably well. So the model is clearly learning useful biology.
It just does not seem to capture specific pairwise interaction effects through this zero-shot scoring approach.
That distinction matters.
Because it suggests that current protein language models may be good at asking:
“How harmful is this mutation on its own?”
But still poor at asking:
“How does this mutation change the meaning of another one?”
And that second question is where epistasis begins.
The remaining epistasis is actually more interesting
Once the artifact is removed, the signal that remains becomes much more interpretable.
Nearby positions in sequence show stronger epistasis than distant ones. That suggests that real epistasis is tied to structural proximity, not just statistical distortion.
The cleaned data also reveals epistatic hub positions. In other words, some residues seem to participate in many more non additive interactions than others.
That is a much more satisfying scientific story:
Not “epistasis is everywhere.”
But rather:
specific residues interact in structurally meaningful ways, and we can finally start seeing them once we stop confusing artifacts for biology.
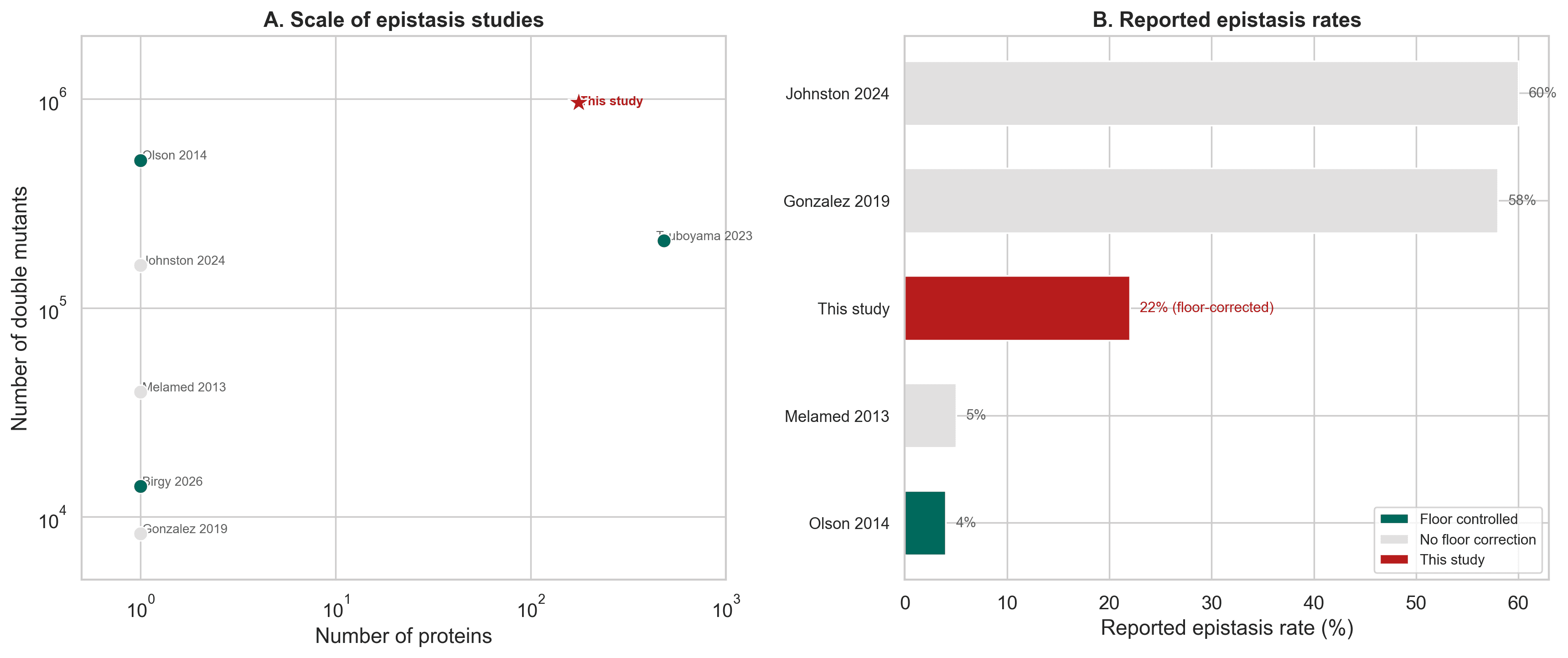
Figure 7. Nearby residues tend to interact more strongly. Once noise is removed, a clearer structure-related signal emerges.
Some of the most interesting patterns only appear after cleaning the data
This was one of the most exciting parts of the analysis.
Once we focus on mild mutations, previously hidden patterns emerge:
Aromatic pairs are more epistatic
When both mutations introduce aromatic residues, the mean epistasis becomes much stronger. That is consistent with stacking interactions and tight packing effects.
Conservative mutations can be more epistatic than radical ones
This is counterintuitive at first, but it makes sense if subtle changes preserve enough function for fine-grained pairwise effects to matter.
Nearby mutations flip outcomes more often
Sign epistasis is stronger when residues are closer in sequence.
N-terminal residues are more epistatic
That may reflect the importance of early folding events and how initial structural organization shapes the rest of the protein.
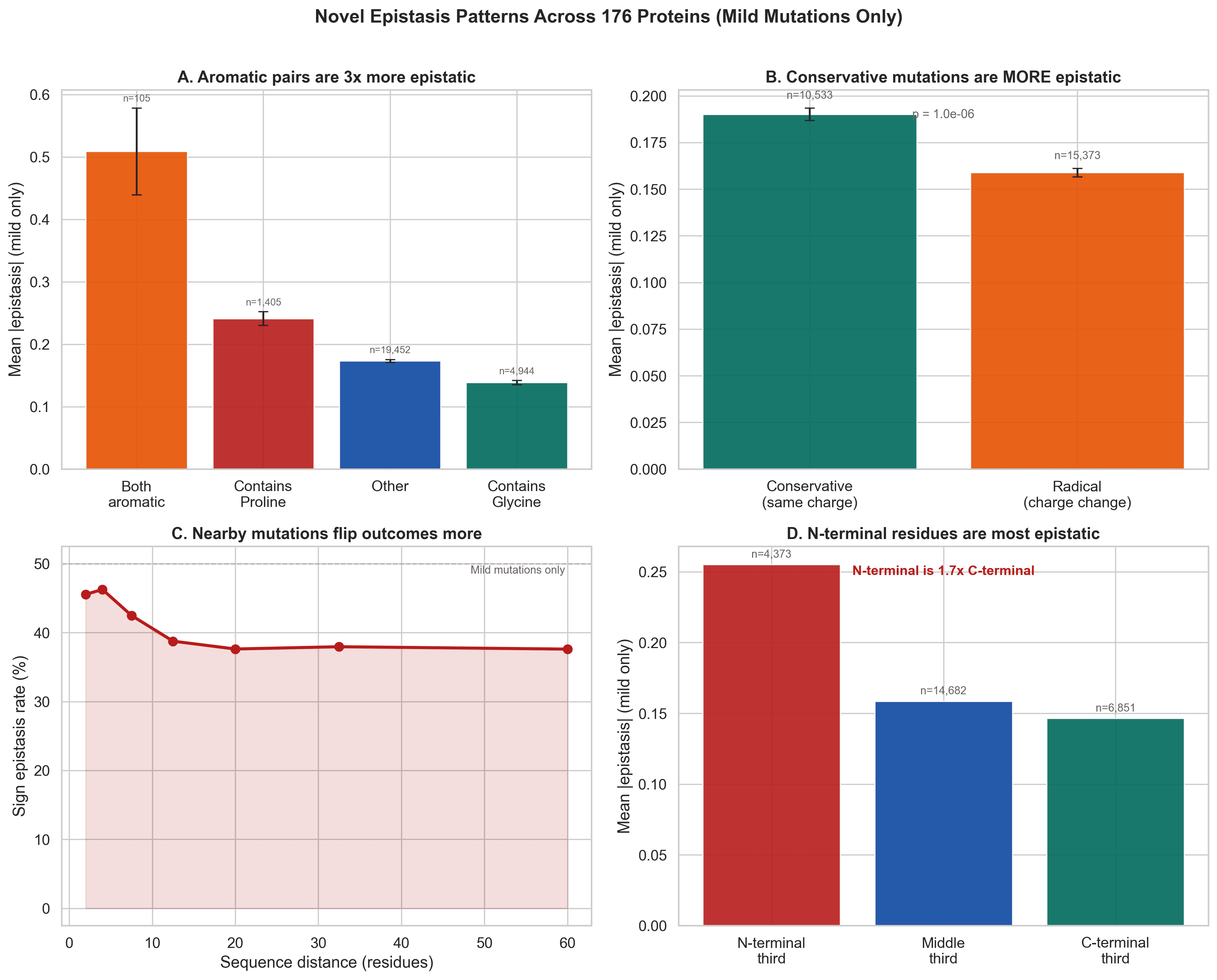
Figure 8. Mild-mutation analysis uncovers specific patterns that were hidden in the raw data, including aromatic effects, conservative substitution effects, sequence-distance dependence, and N-terminal enrichment.
The synergy myth also gets weaker
A common assumption in protein evolution is that positive or synergistic epistasis dominates.
Raw data seems to support that.
But again, the floor effect explains most of that story.
For mild mutations, the balance between synergistic and antagonistic effects becomes much more even.
So the widespread belief that protein evolution is dominated by positive synergy looks much shakier once measurement artifacts are controlled.
Why this matters beyond one benchmark
This is not just a technical correction.
It changes how we should think about protein machine learning.
If epistasis benchmarks are inflated by floor effects, then models may look better than they really are. They may not be learning mutation interaction. They may simply be learning when proteins are already close to the floor.
That matters for anyone building models for:
protein engineering
resistance prediction
directed evolution
multi-mutation design
fitness landscape modeling
Because if a model cannot distinguish artifact from interaction, it may fail exactly where biology becomes most interesting.
The final impact
This work does three important things.
1. It gives the field a cleaner baseline
The real epistasis rate appears to be much closer to 22%, not the dramatic ~60% often implied by raw analyses.
2. It sharpens the scientific question
The challenge is not whether epistasis exists. It clearly does. The challenge is whether we can isolate the specific, structurally meaningful part of it.
3. It raises the bar for protein language models
ESM-2 performs well on single mutation effects, but this analysis suggests that zero-shot scoring does not solve pairwise interaction prediction.
That is a valuable negative result.
Negative results like this save the field time. They stop us from confusing confidence with progress.
And they point us toward what needs to happen next.
So what comes next?
Epistasis prediction remains open. Neither additive models nor current protein language model scoring approaches solve it.
A few directions now look especially important:
Structure-aware models
If real epistasis is tied to proximity, residue hubs, and physical constraints, then models that reason directly over structure should have an advantage.
Models trained on combinatorial data
Single-mutation learning is not the same as interaction learning. If we care about epistasis, we may need objectives trained directly on multi-mutant outcomes.
Better decomposition of global vs specific effects
Separating floor-like nonlinearities from genuine residue-residue interaction will be essential for honest benchmarking and model development.
Cleaner benchmarks
Before claiming a model predicts epistasis, we should first ask whether the benchmark is rewarding true interaction prediction or merely artifact detection.
The most interesting thing about this study is not that a model failed.
It is that once the noise is removed, the biology gets clearer.
The easy story was:
“Epistasis is everywhere, and ESM-2 can predict it.”
The more useful story is:
“A lot of apparent epistasis is measurement geometry. But the epistasis that remains is real, structured, and still unsolved.”
That is a better place for the field to be.
Less myth. More mechanism.
And a much sharper target for the next generation of protein models.
Book a 20-Minute Demo
Sign up free for unlimited Overview runs — summary, sequence-based analysis, homology search. For the full Characterization — PTMs, binding sites, stability variants, construct design — book a demo and we'll run your target live.
