Blog
Orbion Team
Why Your Protein Precipitates During Buffer Exchange

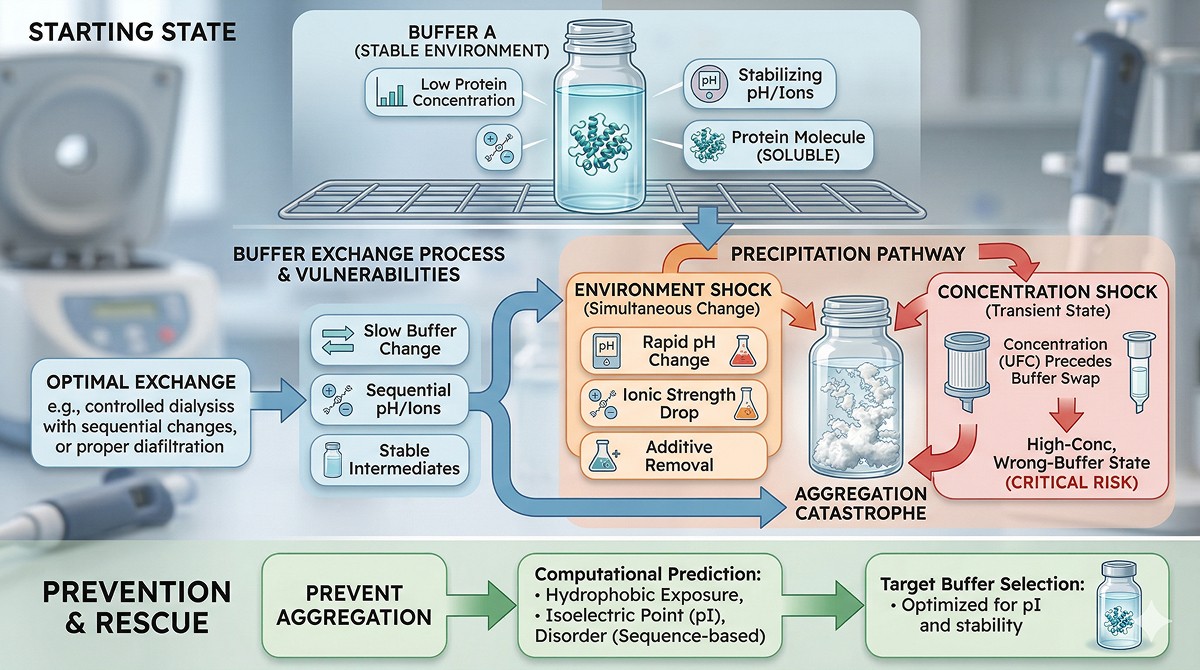
You spent three days purifying your protein. Size exclusion gave a beautiful monodisperse peak. You measured the concentration: 8 mg/mL, perfect for crystallization trials. Then you dialyzed overnight into your assay buffer. The next morning, the dialysis cassette is cloudy. You centrifuge. Half your protein is in the pellet. The other half has shifted to a higher molecular weight species on SEC. Three days of work, gone in one buffer change.
Buffer exchange is supposed to be the easy step—the one between "I have pure protein" and "I have useful protein." Instead, it's a graveyard for unprepared researchers.
Key Takeaways
Buffer exchange precipitation is a concentration + environment problem, not a protein quality problem—the protein was fine until you changed its conditions
The most dangerous variables are pH, ionic strength, and removal of stabilizing additives—changing all three simultaneously (as dialysis often does) is a recipe for aggregation
Concentration during exchange kills more protein than the exchange itself: diafiltration concentrates before full buffer equilibration, creating a transient high-concentration, wrong-buffer state
Predictable from sequence: proteins with high hydrophobic surface exposure, low net charge near target pH, or disorder-prone regions are at highest risk
Prevention is easier than rescue: computational assessment of aggregation propensity and isoelectric point before choosing your target buffer saves protein and time
The Physics of Precipitation During Buffer Exchange
What's Actually Happening
Protein solubility depends on a delicate balance of forces:
Electrostatic repulsion between like-charged molecules (keeps them apart)
Hydrophobic attraction between exposed nonpolar patches (pulls them together)
Solvation by water and co-solutes (stabilizes the folded state)
Conformational stability (keeps hydrophobic core buried)
Buffer exchange disrupts this balance. When you change pH, ionic strength, or remove stabilizers, you shift the equilibrium—sometimes past the solubility limit.
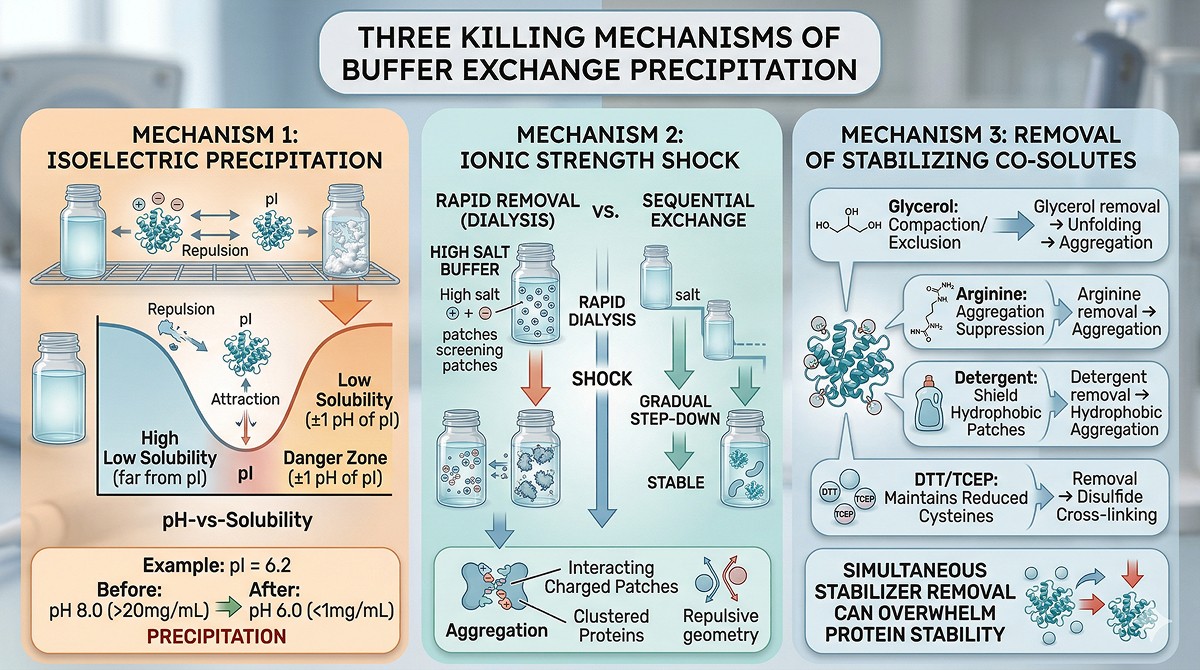
The Three Killing Mechanisms
Mechanism 1: Isoelectric precipitation
Every protein has an isoelectric point (pI) where its net charge is zero. At the pI, electrostatic repulsion between molecules vanishes, and attractive forces (hydrophobic, van der Waals) dominate.
If your target buffer pH is within ±1 unit of the protein's pI, you're in the danger zone
At the pI, solubility can drop 10–100x compared to pH values 2 units away
This is the most common cause of buffer exchange precipitation
Example: Your protein has a pI of 6.2. It was purified in Tris pH 8.0 (net charge ~ –8). You dialyze into MES pH 6.0 (net charge ~ –1). Solubility drops from >20 mg/mL to <1 mg/mL. Precipitation.
Mechanism 2: Ionic strength shock
Proteins in high-salt buffers (post-ion exchange or HIC) have their charged patches screened. When you remove salt rapidly:
Charged patches on adjacent molecules interact (attractive or repulsive, depending on geometry)
If attractive patches align, aggregation occurs
Some proteins require minimum ionic strength for solubility—dropping below it causes "salting in" failure
Conversely, proteins purified in low-salt conditions can precipitate when moved to high salt if they rely on electrostatic repulsion for solubility.
Mechanism 3: Removal of stabilizing co-solutes
Many purification buffers contain stabilizers that get removed during exchange:
Co-solute | What It Does | What Happens When Removed |
|---|---|---|
Glycerol (5–10%) | Preferential exclusion, compacts protein | Unfolding, aggregation |
Arginine (50–500 mM) | Suppresses aggregation | Aggregation of partially unfolded intermediates |
Detergent (0.01–0.1%) | Shields hydrophobic patches | Hydrophobic aggregation |
Imidazole (residual) | Weakly stabilizing for some proteins | Minor, but can contribute |
DTT/TCEP | Maintains reduced cysteines | Disulfide-mediated aggregation |
EDTA | Chelates metals | Metal-catalyzed oxidation and cross-linking |
Removing multiple stabilizers simultaneously (as happens in dialysis into a "clean" buffer) can push marginally stable proteins over the edge.
The Concentration Trap
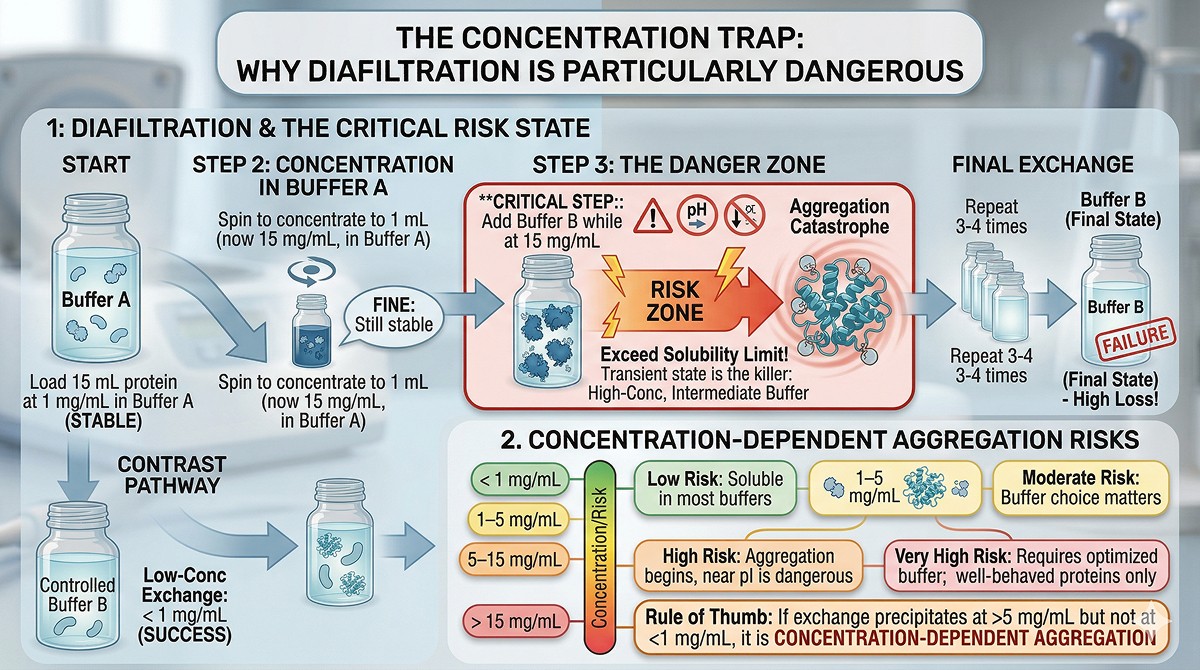
Why Diafiltration Is Particularly Dangerous
Centrifugal concentrators (Amicon, Vivaspin) are the most common buffer exchange method. But they have a critical problem: they concentrate the protein before the buffer is fully exchanged.
What happens during diafiltration:
You load 15 mL of protein at 1 mg/mL in Buffer A
You spin to concentrate to 1 mL (now 15 mg/mL, still mostly in Buffer A)
You dilute with 15 mL Buffer B
You spin again to concentrate
You repeat 3–4 times
The danger: At step 2, your protein is at 15 mg/mL in Buffer A. That's fine—it was stable in Buffer A. But at step 3, you've added Buffer B while the protein is at 15 mg/mL. If the protein is less soluble in Buffer B, you just exceeded its solubility limit at 15 mg/mL before you've done enough rounds to actually reach Buffer B conditions.
The transient state is the killer: Your protein passes through a high-concentration, intermediate-buffer condition that may be far worse than either pure Buffer A or pure Buffer B.
Concentration-Dependent Aggregation
Concentration | Risk Level | Notes |
|---|---|---|
< 1 mg/mL | Low | Most proteins are soluble at low concentration in most buffers |
1–5 mg/mL | Moderate | Buffer composition starts to matter |
5–15 mg/mL | High | Many proteins begin aggregating, especially near pI |
> 15 mg/mL | Very high | Only well-behaved proteins survive; requires optimized buffer |
Rule of thumb: If your protein precipitates during buffer exchange at >5 mg/mL but not at <1 mg/mL, the problem is concentration-dependent aggregation, not buffer incompatibility.
Predicting Which Proteins Will Precipitate
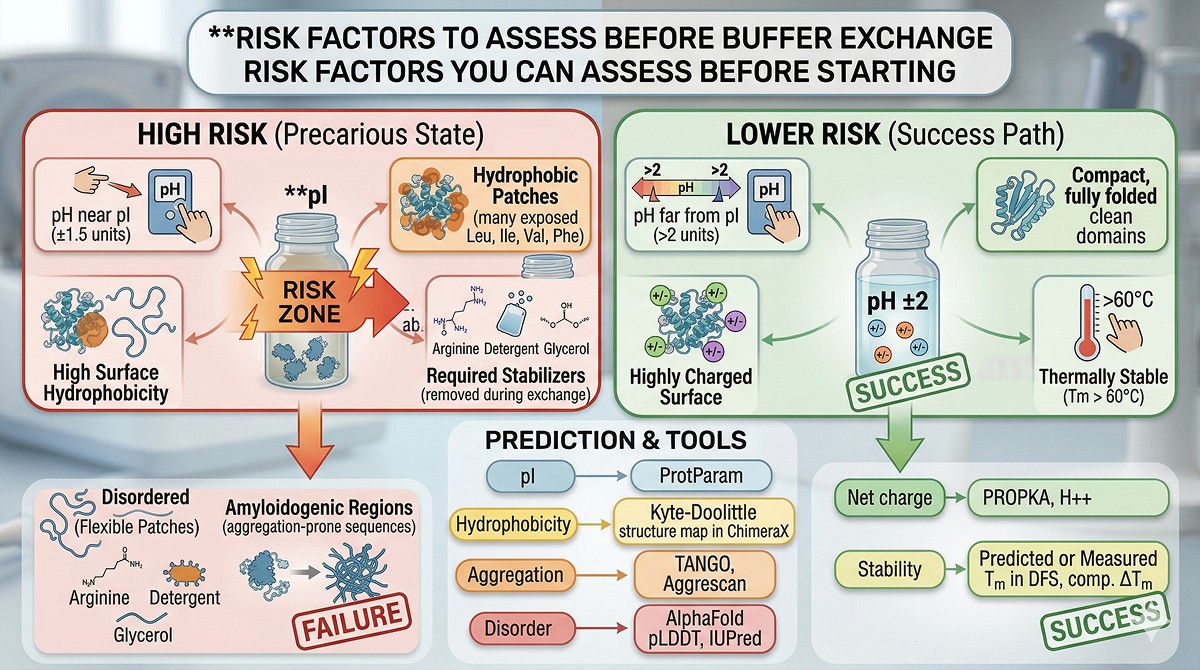
Risk Factors You Can Assess Before Starting
Not all proteins are equally vulnerable. These factors increase precipitation risk:
High risk:
pI within ±1.5 units of target buffer pH
High surface hydrophobicity (many exposed Leu, Ile, Val, Phe patches)
Predicted disordered regions (exposed aggregation-prone sequences)
Known aggregation-prone regions (amyloidogenic stretches)
Proteins that required detergent, arginine, or glycerol during purification
Lower risk:
Target buffer pH >2 units from pI (net charge provides electrostatic repulsion)
Highly charged surface (Asp, Glu, Lys, Arg-rich)
Compact, fully folded domains with minimal exposed hydrophobicity
Thermally stable (Tm > 60°C)
Computational Assessment
Property | How to Assess | Tool |
|---|---|---|
Isoelectric point | Calculate from sequence | ExPASy ProtParam, any sequence tool |
Surface hydrophobicity | Map Kyte-Doolittle onto structure | PyMOL, ChimeraX, or structural viewers |
Aggregation propensity | Sequence-based prediction | TANGO, Aggrescan, Zyggregator |
Disorder regions | pLDDT from AlphaFold, IUPred | AlphaFold DB, disorder predictors |
Net charge at target pH | Henderson-Hasselbalch for all titratable groups | PROPKA, H++ |
Thermal stability | Predicted or measured Tm | DSF, computational ΔTm |
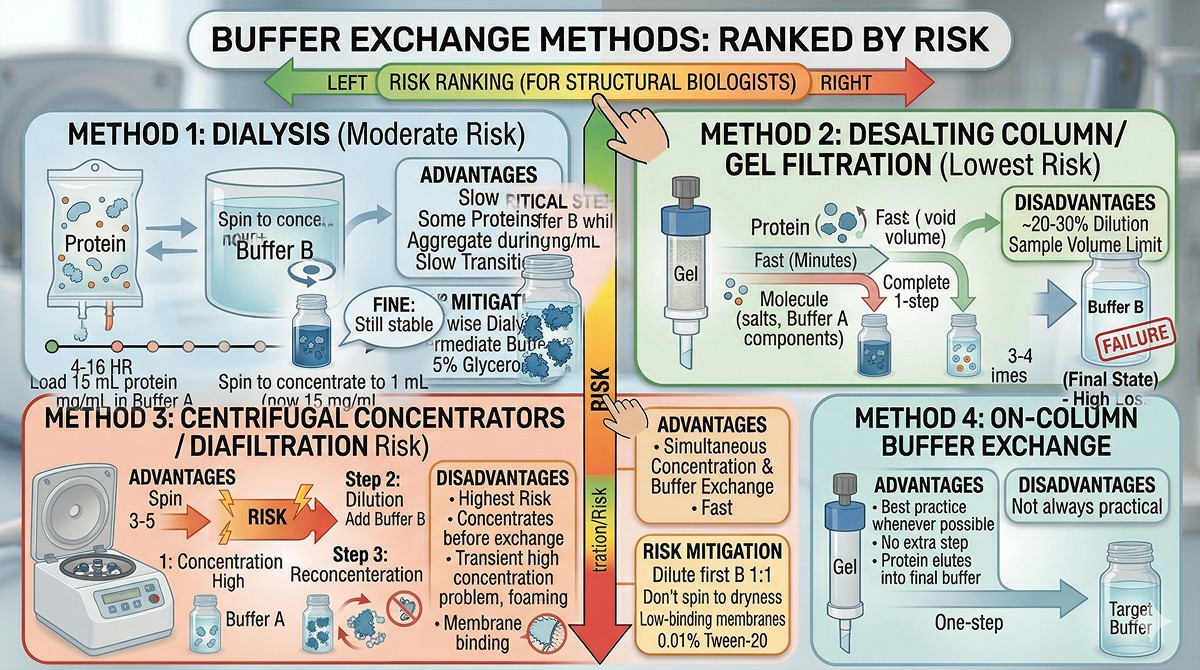
Buffer Exchange Methods: Ranked by Risk
Method 1: Dialysis (Moderate Risk)
How it works: Protein in a semi-permeable membrane equilibrates with a large volume of target buffer over 4–16 hours.
Advantages:
Gentle (no concentration step)
Complete buffer exchange if done with 2–3 buffer changes
Works at any concentration
Disadvantages:
Slow (hours to overnight)
Protein concentration doesn't change (can't concentrate simultaneously)
Some proteins aggregate during the slow pH/salt transition
Dilute samples lose protein to membrane binding
Risk mitigation:
Use stepwise dialysis: intermediate buffer → final buffer (avoids sharp transitions)
Keep protein at moderate concentration (<5 mg/mL during exchange)
Add 5% glycerol to the dialysis buffer if protein is marginally stable
Method 2: Desalting Column / Gel Filtration (Lowest Risk)
How it works: Protein passes through a gel filtration matrix (e.g., Sephadex G-25, PD-10, HiTrap Desalting) pre-equilibrated in target buffer. Small molecules (salt, buffer components) are retarded; protein flows through in the void volume.
Advantages:
Fast (minutes)
Complete buffer exchange in one step
No concentration effect (protein comes out at roughly the same concentration)
Minimal time in intermediate conditions
Disadvantages:
~20–30% dilution (void volume > sample volume)
Sample volume limited by column capacity
Multiple runs needed for large volumes
This is the safest method for buffer exchange. If your protein is precipitation-prone, use desalting columns.
Method 3: Centrifugal Concentrators / Diafiltration (Highest Risk)
How it works: Protein solution is concentrated through a molecular weight cutoff membrane, then diluted with target buffer and reconcentrated. Repeated 3–5 times.
Advantages:
Simultaneous concentration and buffer exchange
Fast
Scalable
Disadvantages:
Concentrates before exchanging (the transient high-concentration problem)
Membrane binding (hydrophobic proteins stick to PES/cellulose membranes)
Foaming at the membrane surface can denature protein
Uneven concentration at the membrane surface (local concentration much higher than bulk)
Risk mitigation:
Dilute the first addition of target buffer 1:1 with original buffer (gradual transition)
Don't spin to dryness—keep a minimum 2 mL above the membrane
Use low-binding membranes (regenerated cellulose > PES for sticky proteins)
Add 0.01% Tween-20 to reduce membrane binding
Method 4: On-Column Buffer Exchange (Low Risk)
How it works: If your last purification step is a column (SEC, ion exchange), simply equilibrate the column in your target buffer.
Advantages:
No extra step—the purification IS the buffer exchange
Protein elutes directly into target buffer
No intermediate states
Disadvantages:
Not always practical (SEC in target buffer may change elution behavior)
May need to re-optimize elution conditions
Best practice whenever possible: Plan your purification so the last column step delivers protein in (or close to) your final buffer.
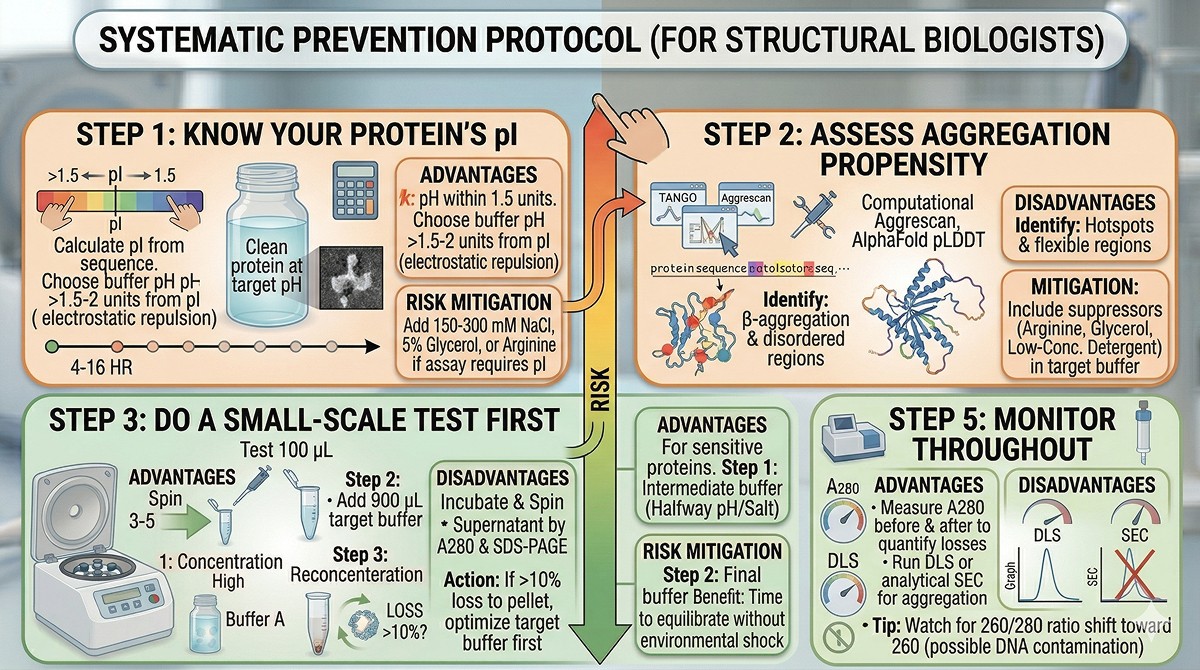
The Systematic Prevention Protocol
Step 1: Know Your Protein's pI
Calculate the pI from the amino acid composition. If your target buffer pH is within 1.5 units of pI, you're at risk.
What to do:
Choose a buffer pH at least 1.5–2 units away from pI
If you can't (assay requires specific pH), add 150–300 mM NaCl to screen charges
Or add 5% glycerol or 200 mM arginine to suppress aggregation
Step 2: Assess Aggregation Propensity
Use computational tools to predict aggregation-prone regions:
TANGO predicts β-aggregation propensity
Aggrescan identifies aggregation hotspots from sequence
AlphaFold pLDDT flags disordered regions that expose aggregation-prone sequences
If your protein has high predicted aggregation propensity, include aggregation suppressors (arginine, glycerol, low-concentration detergent) in your target buffer.
Step 3: Do a Small-Scale Test First
Never exchange your entire prep without testing first.
Take 100 µL of purified protein
Add 900 µL of target buffer (10x dilution)
Incubate at 4°C for 1 hour
Centrifuge at 16,000g for 10 minutes
Check supernatant by A280 and SDS-PAGE
If you see >10% loss to the pellet, the target buffer needs optimization before you commit your full prep.
Step 4: Exchange Gradually
If your protein is sensitive:
Step 1: Exchange into an intermediate buffer (halfway between current and target pH/salt)
Step 2: Exchange into the final buffer
Each step gives the protein time to equilibrate without a drastic environmental shift
Step 5: Monitor Throughout
Measure A280 before and after to quantify losses
Run DLS or analytical SEC after exchange to check for aggregation
If the A280 ratio (260/280) shifts toward 260, you may have nucleic acid contamination from aggregated protein binding DNA
Rescue Strategies: When Precipitation Has Already Happened
If You Catch It Early (Slight Turbidity)
Centrifuge gently (5,000g, 5 min) to remove large aggregates
Add stabilizer to the supernatant: glycerol to 10%, or arginine to 200 mM, or NaCl to 300 mM
Check concentration and activity of the rescued fraction
Move fast—partially aggregated protein continues to aggregate
If It's Heavily Precipitated
Centrifuge and save the pellet
Try resolubilizing in high-salt buffer (500 mM NaCl, pH 2 units from pI)
If it resolubilizes, the aggregation was reversible (likely electrostatic)
If it doesn't, the aggregation is irreversible (hydrophobic collapse, likely denatured)
Resolubilized protein must be checked by SEC—monomeric fraction may still be usable
When It's Not Recoverable
If the protein is irreversibly aggregated:
The problem is likely conformational instability, not just buffer incompatibility
Consider adding stabilizers to ALL future buffers from lysis onward
Redesign the construct (remove disordered regions that expose aggregation patches)
Try a different expression system or fusion partner
Buffer Optimization Quick Reference
The Universal Safe Buffer
When you don't know what your protein likes, start here:
50 mM Tris pH 8.0, 150 mM NaCl, 5% glycerol, 1 mM DTT
This works for most soluble, cytoplasmic proteins because:
pH 8.0 is above most protein pIs (average pI for human cytoplasmic proteins is ~7.4)
150 mM NaCl screens charges without salting out
5% glycerol suppresses aggregation
1 mM DTT prevents oxidative cross-linking
pH Recommendations by Protein pI
Protein pI | Recommended Buffer pH | Rationale |
|---|---|---|
< 5.5 | 7.0–8.0 (well above pI) | Strong negative charge |
5.5–7.0 | 8.0–8.5 | Avoid the pI zone |
7.0–8.0 | 6.0 or 9.0 (flank the pI) | Must choose one side |
8.0–9.0 | 6.5–7.5 (below pI) | Positive charge at lower pH |
> 9.0 | 7.0–8.0 (well below pI) | Strong positive charge |
Additives That Prevent Precipitation
Additive | Concentration | Mechanism | When to Use |
|---|---|---|---|
NaCl | 150–500 mM | Charge screening | Near pI, charged surfaces |
Glycerol | 5–10% | Preferential exclusion | General stabilizer |
L-Arginine | 200–500 mM | Aggregation suppression | Exposed hydrophobic patches |
Sucrose/Trehalose | 5–10% | Preferential exclusion | Thermally unstable proteins |
Tween-20 | 0.01–0.05% | Shields hydrophobic surfaces | Membrane proteins, hydrophobic proteins |
TCEP (1–5 mM) | 1–5 mM | Reduces disulfides | Proteins with free cysteines |
The Bottom Line
Problem | Cause | Prevention |
|---|---|---|
Precipitation during dialysis | pH transition through pI | Stepwise dialysis, check pI first |
Loss during diafiltration | Transient high-concentration in wrong buffer | Use desalting column instead, or dilute gradually |
Aggregation after removing glycerol/arginine | Loss of stabilizing co-solute | Keep stabilizers in target buffer |
Cloudiness at high concentration | Exceeding solubility limit | Exchange at low concentration, then concentrate |
Disulfide-mediated aggregation | Loss of reducing agent | Include TCEP in target buffer |
The simplest rule: know your protein's pI, never exchange into a buffer near it, and always do a small-scale test first. These three habits prevent 90% of buffer exchange disasters.

Designing Buffer-Safe Workflows with Orbion
Orbion can help you anticipate buffer exchange problems before they happen. AstraUNFOLD predicts per-residue disorder probability and amyloidogenicity—flagging regions that expose aggregation-prone sequences during partial unfolding. The Bench module generates purification and formulation protocols that account for the protein's predicted properties, including buffer pH recommendations relative to the calculated pI and stabilizer suggestions based on predicted aggregation risk.
Combined with Construct Design for removing disordered regions that drive aggregation, you can design a construct and buffer strategy together—addressing the root cause rather than treating the symptoms after your protein has already crashed out of solution.
References
Arakawa T & Tsumoto K. (2003). The effects of arginine on refolding of aggregated proteins: not facilitate refolding, but suppress aggregation. Biochemical and Biophysical Research Communications, 304(1):148-152. Link
Bondos SE & Bhatt A. (2001). Concentration in protein science: not a trivial problem. Focus, 21(1):7-10. PMC7115950
Arakawa T, et al. (2007). Suppression of protein interactions by arginine: a proposed mechanism of the arginine effects. Biophysical Chemistry, 127(1-2):1-8. Link
Timasheff SN. (1998). Control of protein stability and reactions by weakly interacting cosolvents: the simplicity of the complicated. Advances in Protein Chemistry, 51:355-432. Link
Golovanov AP, et al. (2004). A simple method for improving protein solubility and long-term stability. Journal of the American Chemical Society, 126(29):8933-8939. Link
Fernandez-Escamilla AM, et al. (2004). Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nature Biotechnology, 22(10):1302-1306. Link
Conchillo-Solé O, et al. (2007). AGGRESCAN: a server for the prediction and evaluation of "hot spots" of aggregation in polypeptides. BMC Bioinformatics, 8:65. PMC1828742
Shire SJ, et al. (2004). Challenges in the development of high protein concentration formulations. Journal of Pharmaceutical Sciences, 93(6):1390-1402. Link
Kramer RM, et al. (2012). Toward a molecular understanding of protein solubility: increased negative surface charge correlates with increased solubility. Biophysical Journal, 102(8):1907-1915. PMC3328702
Cromwell MEM, et al. (2006). Protein aggregation and bioprocessing. The AAPS Journal, 8(3):E572-E579. PMC2750999
Book a 20-Minute Demo
Sign up free for unlimited Overview runs — summary, sequence-based analysis, homology search. For the full Characterization — PTMs, binding sites, stability variants, construct design — book a demo and we'll run your target live.
