Blog
Orbion Team
Protein Language Models Explained for Bench Scientists

Your colleague mentions that their new prediction tool uses "ESM-2 embeddings." Your PI asks whether you should use "protein language models" for variant analysis. A preprint claims that "masked language modeling" predicts mutation effects better than Rosetta. You nod along in the lab meeting, but privately you have no idea what any of this actually means, whether it's hype, or whether you should care.
You should care. Protein language models are quietly reshaping how we predict structure, function, and mutation effects—but the field has done a remarkably poor job explaining them to the bench scientists who actually use proteins.
Key Takeaways
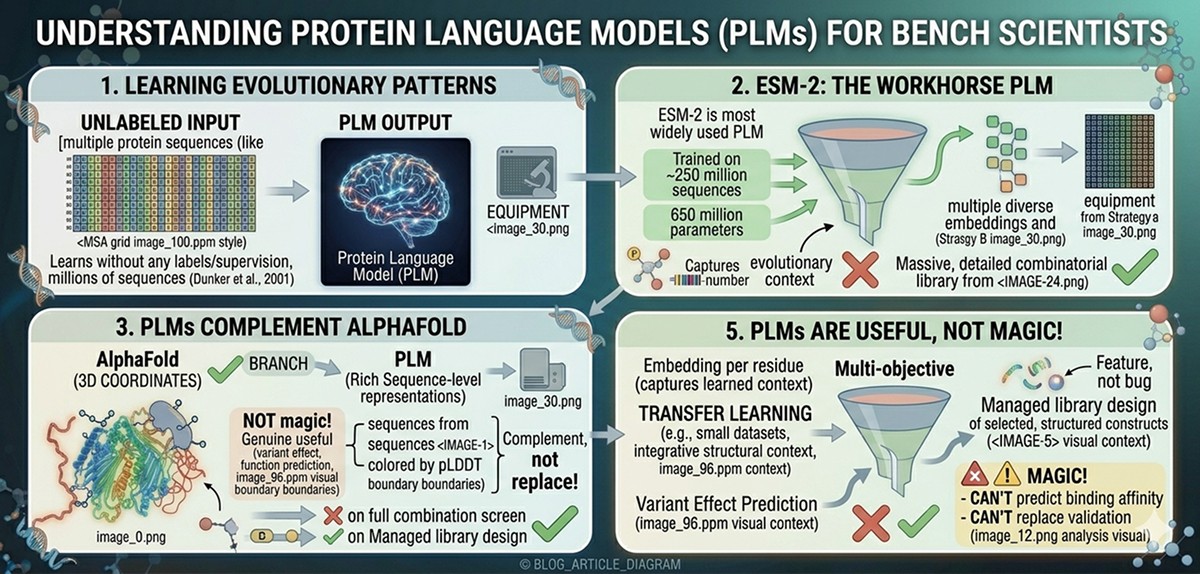
Protein language models (PLMs) learn evolutionary patterns from millions of protein sequences without any labels or supervision
ESM-2 is the most widely used PLM: 650 million parameters trained on ~250 million sequences, producing per-residue "embeddings" that encode evolutionary and structural information
PLMs complement AlphaFold, not replace it: AlphaFold predicts 3D coordinates; PLMs provide rich sequence-level representations useful for variant scoring, function prediction, and engineering
"Embeddings" are high-dimensional fingerprints: a 1,280-number vector per residue that captures what the model learned about that position's evolutionary context
PLMs are genuinely useful but not magic: they excel at variant effect prediction and transfer learning for small datasets, but can't predict binding affinity or replace experimental validation
The Core Analogy: Protein Sequences as Language
Why the Analogy Works
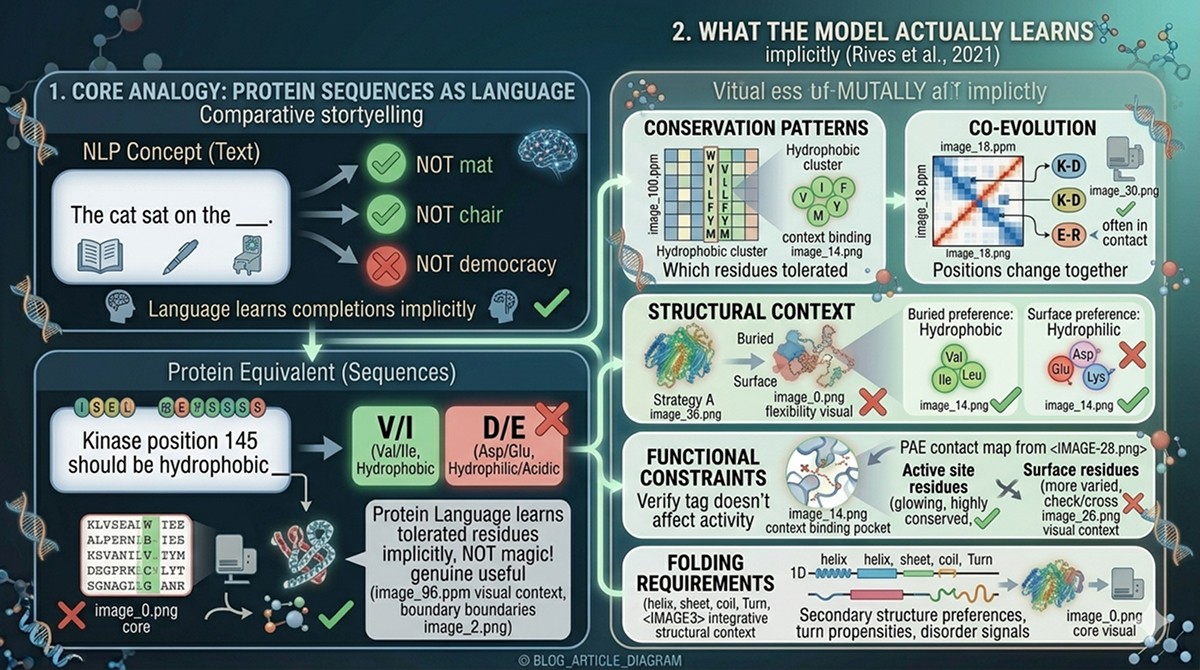
Natural language processing (NLP) revolutionized text understanding by treating words as elements in a structured language with grammar and meaning. The insight behind protein language models is that protein sequences behave similarly:
NLP Concept | Protein Equivalent |
|---|---|
Words | Amino acids (20-letter alphabet) |
Sentences | Protein sequences |
Grammar | Physical and evolutionary constraints on sequence |
Meaning | Structure, function, interactions |
Dialects | Protein families with conserved patterns |
Typos | Deleterious mutations |
Synonyms | Conservative substitutions (V↔I, D↔E) |
The key parallel: Just as a language model learns that "The cat sat on the ___" should be completed with "mat" or "chair" (not "democracy"), a protein language model learns that position 145 in a kinase should be a hydrophobic residue (not an aspartate)—without being told anything about kinases, hydrophobicity, or protein folding.
What the Model Actually Learns
When you train a language model on billions of words of English, it learns grammar, semantics, and world knowledge—all implicitly. Similarly, when you train a protein language model on hundreds of millions of protein sequences, it implicitly learns (Rives et al., 2021):
Conservation patterns: Which residues are tolerated at each position in a protein family
Co-evolution: Which positions change together (often because they're in contact)
Structural context: Buried positions prefer hydrophobic residues; surface positions prefer hydrophilic ones
Functional constraints: Active site residues are more conserved than surface residues
Folding requirements: Secondary structure preferences, turn propensities, disorder signals
None of this is programmed in. The model discovers it from data alone.
What Is a Protein Language Model?
Self-Supervised Learning on Sequences
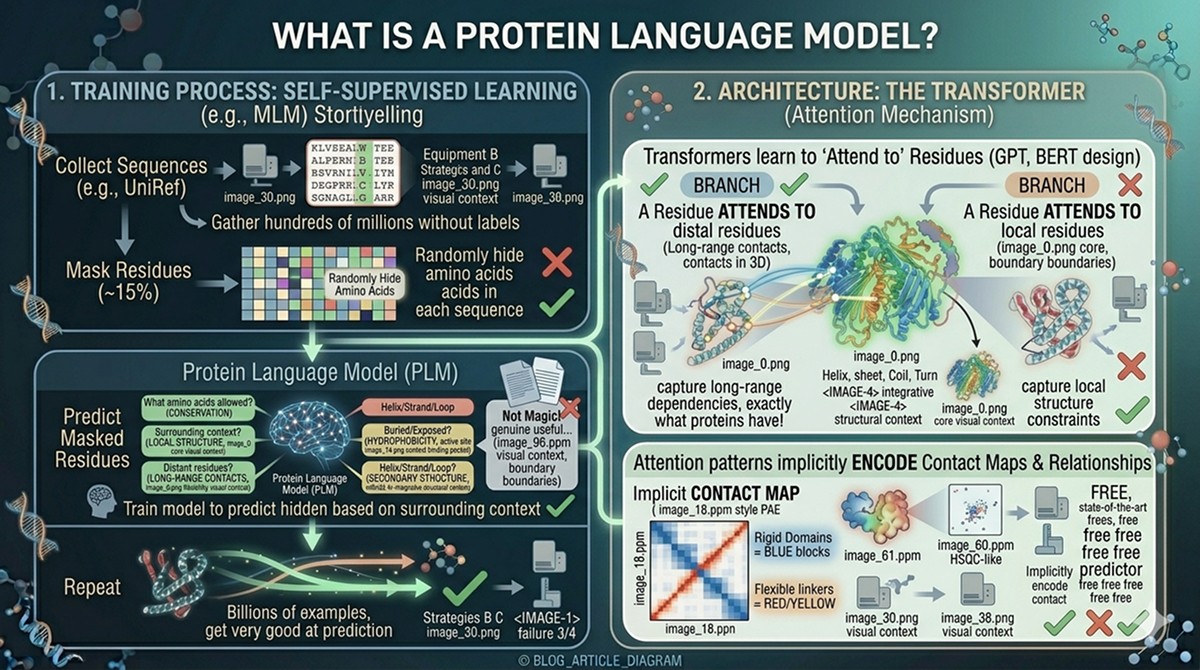
The central idea is self-supervised learning: you train the model on a vast amount of data without manual labels.
The training process:
Collect sequences: Gather hundreds of millions of protein sequences from databases like UniRef
Mask residues: Randomly hide ~15% of amino acids in each sequence
Predict the masked residues: Train the model to predict what was hidden, based on the surrounding context
Repeat: Process billions of examples until the model gets very good at prediction
This is called masked language modeling (MLM)—the same approach that powers BERT in NLP.
What the model learns from this simple task:
To predict a masked residue accurately, the model must understand:
What amino acids are allowed at this position (conservation)
How neighboring residues constrain the choice (local structure)
How distant residues influence this position (long-range contacts, coevolution)
Whether this position is buried or exposed (hydrophobicity patterns)
Whether this position is in a helix, strand, or loop (secondary structure preferences)
All of this emerges from the single task of predicting masked residues.
Architecture: The Transformer
PLMs use the transformer architecture—the same design behind GPT and BERT (Vaswani et al., 2017). The key mechanism is attention: for each residue, the model computes how much every other residue in the sequence matters for understanding it.
Why this matters for proteins:
A residue in the active site might "attend to" other active site residues 200 positions away
Residues in contact in 3D space often attend to each other, even if distant in sequence
The attention patterns implicitly encode contact maps and structural relationships
You don't need to understand the math. The practical takeaway: transformers are architecturally well-suited to proteins because they can capture long-range dependencies in sequences—exactly what proteins have.
ESM-2: The Workhorse Model
What It Is
ESM-2 (Evolutionary Scale Modeling 2) is Meta AI's protein language model (Lin et al., 2023). It's the most widely used PLM in both academic and industrial protein science.
Key specifications:
Property | Value |
|---|---|
Developer | Meta AI (FAIR) |
Parameters | 650 million (most common variant) |
Training data | ~250 million sequences from UniRef |
Input | Single amino acid sequence |
Output | Per-residue embeddings (1,280 dimensions) |
Inference time | Seconds for a typical protein |
Availability | Open source (MIT license) |
Model size variants:
Variant | Parameters | Embedding Dim | Best For |
|---|---|---|---|
ESM-2 (8M) | 8 million | 320 | Quick prototyping |
ESM-2 (35M) | 35 million | 480 | Moderate accuracy, fast |
ESM-2 (150M) | 150 million | 640 | Good balance |
ESM-2 (650M) | 650 million | 1,280 | Best accuracy (standard choice) |
ESM-2 (3B) | 3 billion | 2,560 | Marginal improvement, much slower |
ESM-2 (15B) | 15 billion | 5,120 | Diminishing returns |
The 650M parameter model is the standard. Larger models provide marginal improvements that rarely justify the computational cost.
Why ESM-2 Matters
ESM-2 is important because it provides a universal protein representation that can be used for almost any downstream task:
No alignment needed: Unlike methods that require multiple sequence alignments (MSAs), ESM-2 takes a single sequence as input
Fast: Inference takes seconds, not the minutes/hours required for MSA construction + AlphaFold
Transferable: The same embeddings work for function prediction, stability prediction, variant scoring, and more
Captures evolution implicitly: 250 million sequences' worth of evolutionary information is compressed into the model's weights
What "Embeddings" Actually Are
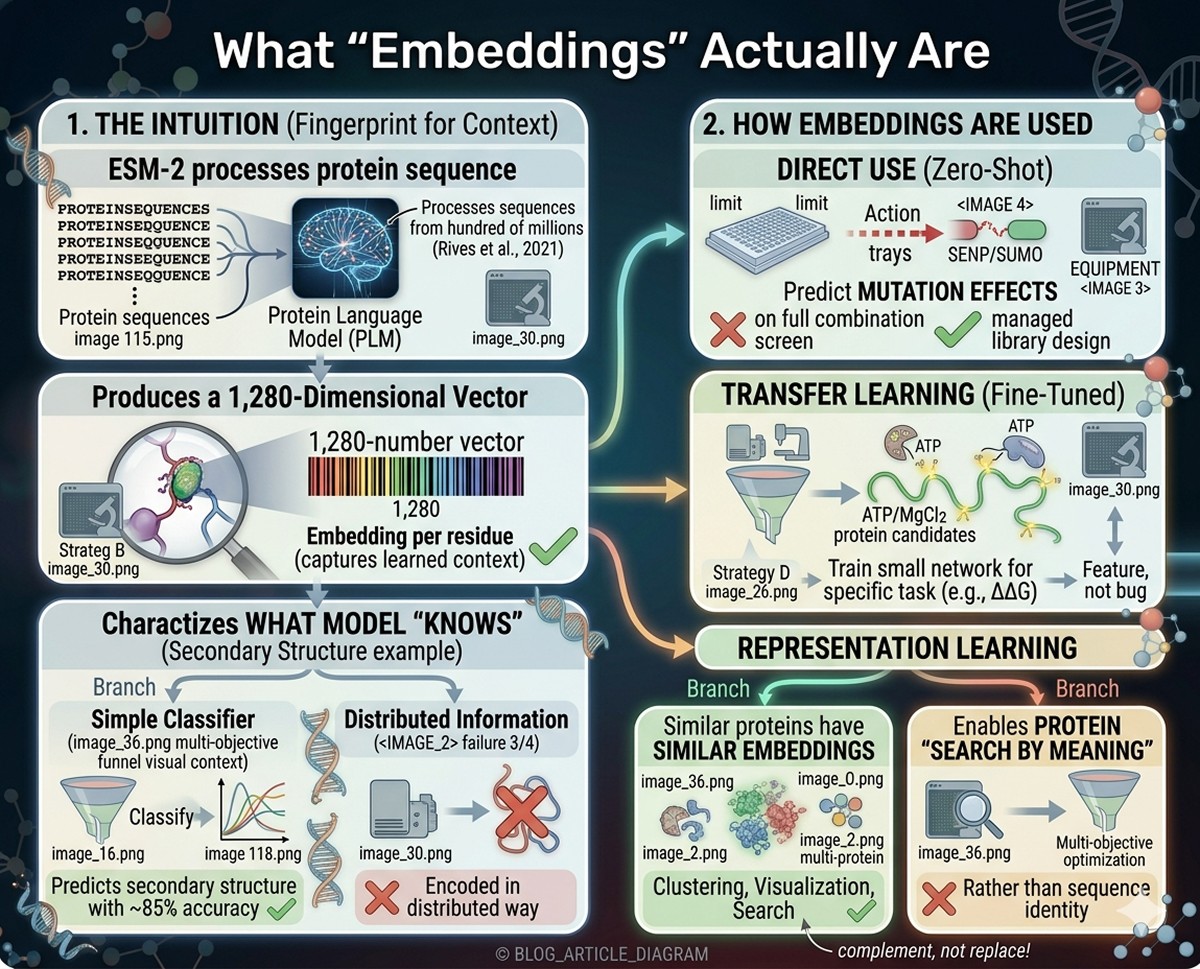
The Intuition
When ESM-2 processes a protein sequence, it produces a 1,280-dimensional vector for each residue. This vector is called an "embedding."
Analogy: Think of it as a fingerprint for each residue's evolutionary and structural context. Just as a fingerprint uniquely identifies a person, an embedding uniquely characterizes what the model "knows" about that position in that protein.
What's encoded in those 1,280 numbers:
Conservation pattern at this position
Secondary structure propensity
Buried vs exposed character
Proximity to functional sites
Co-evolutionary relationships with other positions
Whether the position is in a structured or disordered region
You can't look at the 1,280 numbers and read off "this residue is in a helix." But when you train a simple classifier on top of the embeddings, it can predict secondary structure with ~85% accuracy—the information is there, just encoded in a distributed way.
How Embeddings Are Used
Direct use (zero-shot):
Predict mutation effects by comparing the model's prediction at a position with the actual amino acid
If the model says "this position should be valine" and you mutate it to aspartate, that's likely deleterious
Transfer learning (fine-tuned):
Take the embeddings as input features
Train a small neural network on top for a specific task (e.g., predicting ΔΔG)
Works well even with small training datasets (hundreds of examples)
This is how most practical tools use ESM-2
Representation learning:
Use embeddings as features for clustering, visualization, or search
Similar proteins have similar embeddings
Enables protein "search by meaning" rather than by sequence identity
What "Masked Language Modeling" Means in Practice
The Training Task
During training, ESM-2 sees a sequence like:
For each masked position, the model outputs a probability distribution over all 20 amino acids. The training loss pushes the model to assign high probability to the correct amino acid.
The Practical Implication
After training, you can use this prediction capability directly:
Zero-shot variant scoring:
Feed your wild-type sequence to ESM-2
For each position, the model outputs probabilities for all 20 amino acids
The wild-type amino acid usually has high probability (the model "agrees" with evolution)
A mutation to an amino acid with low probability is likely deleterious
A mutation to an amino acid with moderate probability may be tolerated
The score: log(P_mutant / P_wildtype) gives a variant effect score. Negative = likely deleterious. Positive = possibly beneficial or neutral.
Meier et al. (2021) showed that this zero-shot approach—without ANY training on experimental mutation data—correlates well with deep mutational scanning experiments across multiple proteins.
What PLMs Can Predict (And How Well)
Tasks Where PLMs Excel
Task | How It Works | Accuracy | Reference |
|---|---|---|---|
Secondary structure | Fine-tune on labeled data | ~85% per-residue (Q3) | |
Contact prediction | Attention maps → contacts | Competitive with coevolution methods | |
Variant effect prediction | Zero-shot or fine-tuned | Spearman ρ ~0.4–0.5 with DMS data | |
Subcellular localization | Fine-tune on labeled data | ~90% accuracy | Multiple studies |
Function (GO terms) | Fine-tune on labeled data | AUC ~0.8–0.9 depending on category | Multiple studies |
Disorder prediction | Fine-tune or zero-shot | Comparable to dedicated tools |
Tasks Where PLMs Struggle
Task | Why It's Hard | Better Alternative |
|---|---|---|
Precise 3D structure | Sequence alone lacks 3D information | AlphaFold (uses MSAs + structure modules) |
Absolute binding affinity | Affinity depends on 3D geometry, electrostatics, solvation | Physics-based methods, experimental measurement |
Protein-protein interaction prediction | Requires modeling two sequences jointly | AlphaFold-Multimer, coevolution methods |
Small molecule interactions | PLMs see only amino acids, not ligands | Docking, molecular dynamics |
Post-translational modifications | Some PTMs have weak sequence signals | Combined approaches (sequence + structure) |
Protein dynamics | Single representation, no conformational sampling | Molecular dynamics, NMR |
The Honest Assessment
PLMs are genuinely useful for variant scoring, function prediction, and as input features for downstream models. They are NOT magical—they capture evolutionary patterns, not physics. If a property depends on structural details, electrostatics, or dynamics, PLMs alone won't get you there.
PLMs vs AlphaFold: Complementary, Not Competing
This is the most common source of confusion. PLMs and AlphaFold are different tools that solve different problems:
Feature | Protein Language Models (ESM-2) | AlphaFold2 |
|---|---|---|
Input | Single sequence | Sequence + MSA + templates |
Output | Per-residue embeddings (1,280D vectors) | 3D atomic coordinates |
Speed | Seconds | Minutes to hours |
What it captures | Evolutionary patterns, sequence-level features | 3D structure, spatial relationships |
MSA required? | No | Yes (critical for accuracy) |
Best for | Variant scoring, function prediction, search | Structure prediction, docking, interface analysis |
Limitations | No 3D structure, no physics | Slow, requires MSA, single conformation |
How they work together:
ESM-2 provides fast, universal sequence representations
AlphaFold provides detailed 3D structural models
Many modern tools combine both: ESM-2 embeddings as features + AlphaFold structure as context
ESMFold uses ESM-2 embeddings to predict structure directly (faster than AlphaFold but less accurate)
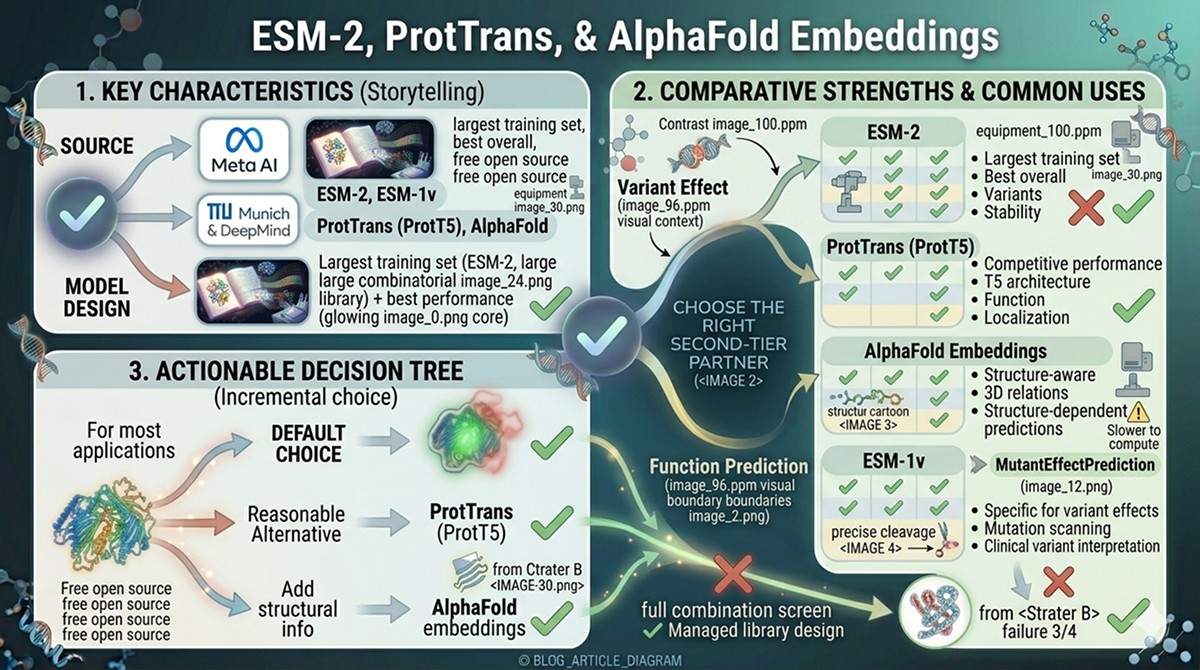
Comparison: ESM-2 vs ProtTrans vs AlphaFold Embeddings
Model | Source | Key Strengths | Common Uses |
|---|---|---|---|
ESM-2 | Meta AI | Largest training set, best overall performance, open source | Variant effect, function prediction, stability |
ProtTrans (ProtT5) | TU Munich | Competitive performance, T5 architecture | Function prediction, localization |
AlphaFold embeddings | DeepMind | Structure-aware, captures 3D relationships | Structure-dependent predictions |
ESM-1v | Meta AI | Specifically designed for variant effects | Mutation scanning, clinical variant interpretation |
For most applications, ESM-2 (650M) is the default choice. It's well-validated, widely supported, and open source. ProtT5 is a reasonable alternative with similar performance. AlphaFold embeddings add structural information but are slower to compute.
How PLMs Are Changing Practical Protein Science
Variant Effect Prediction
The old way: Run Rosetta or FoldX on every possible mutation. Requires a structure. Slow. Physics assumptions may be wrong.
The PLM way: Run ESM-2 zero-shot scoring on every possible mutation. No structure needed. Fast (minutes for entire saturation mutagenesis). Captures evolutionary constraints that physics-based methods miss.
The best way: Combine both. Use PLM scores as one feature alongside ΔΔG from FoldX/Rosetta. Combined models outperform either alone.
Zero-Shot Mutation Scanning
For any protein, you can generate a "mutation landscape" in minutes:
Feed the sequence to ESM-2
For every position, get the model's predictions for all 20 amino acids
Score each possible mutation by log-likelihood ratio
Visualize as a heatmap: positions (rows) × amino acids (columns)
This gives you an immediate sense of which positions are mutationally tolerant and which are constrained—without any experimental data.
Transfer Learning for Small Datasets
The problem: You have 50 experimentally measured ΔTm values for your protein. Training a machine learning model on 50 data points usually gives terrible results.
The PLM solution: Use ESM-2 embeddings as input features. The embeddings already encode evolutionary information from 250 million sequences. Your small dataset just needs to teach the model how to map from those rich features to ΔTm. This is transfer learning, and it works remarkably well.
Studies show that ESM-2-based models can achieve useful accuracy with as few as 50–100 training examples—a fraction of what's needed for models trained from scratch.
Protein Engineering with ProteinMPNN
ProteinMPNN (Dauparas et al., 2022) takes the language model concept further: given a protein backbone structure, it designs sequences that would fold into that structure. This is structure-conditioned language modeling—the model generates "protein language" that is consistent with a given 3D arrangement.
Practical applications:
Redesign protein surfaces for improved solubility
Design sequences for de novo protein structures
Generate diverse sequences for directed evolution starting libraries

The Hype vs Reality Check
What the Preprints Claim vs What Works in the Lab
Claim | Reality | Verdict |
|---|---|---|
"PLMs predict mutation effects" | Zero-shot scores correlate with DMS data (ρ ~0.4–0.5). Useful for ranking, not for precise ΔΔG prediction. | Partially true. Good for prioritization, not precise quantification. |
"ESM-2 understands protein structure" | Embeddings encode structural features implicitly. ESMFold predicts structure but less accurately than AlphaFold. | Partially true. Captures structural patterns but isn't a structure predictor. |
"PLMs replace sequence alignments" | For some tasks (variant scoring), single-sequence PLMs match MSA-based methods. For structure prediction, MSAs are still superior. | Context-dependent. Faster, comparable for some tasks, worse for others. |
"PLMs will design better proteins" | ProteinMPNN designs have high experimental success rates (~50–70% fold correctly). But design is not the same as optimization. | Promising but early. Laboratory validation still required. |
"Bigger models are always better" | ESM-2 (650M) to ESM-2 (15B) shows diminishing returns on most benchmarks. | Mostly false. The 650M model is the sweet spot. |
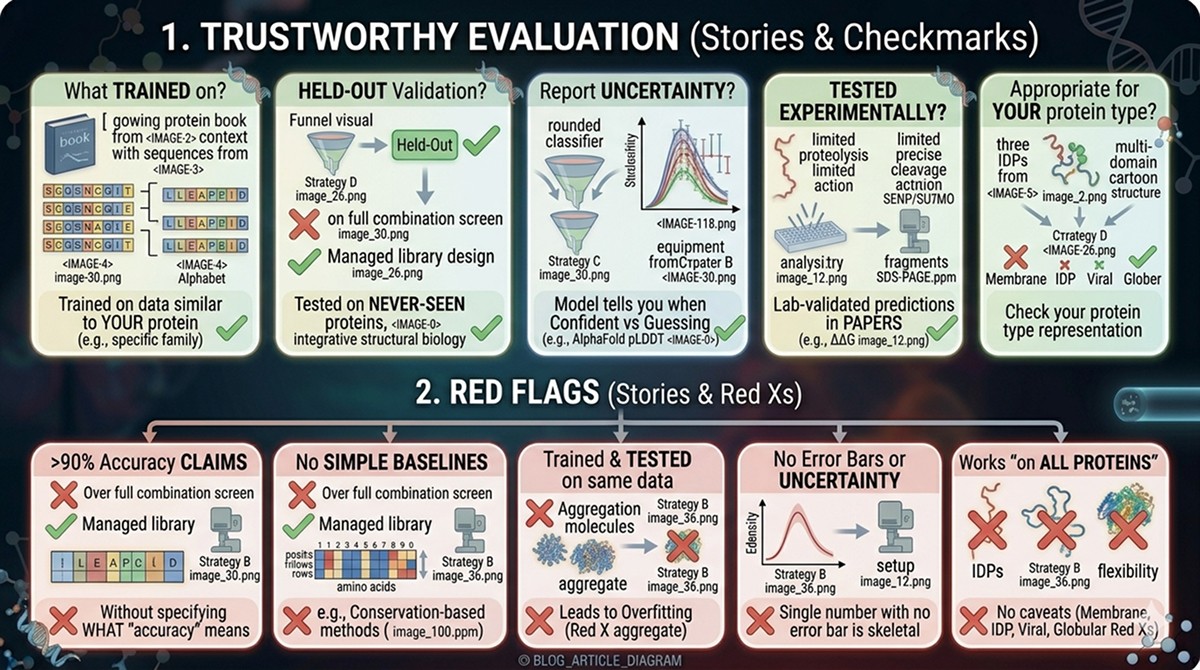
How to Evaluate Whether a PLM-Based Tool Is Trustworthy
Questions to Ask
What was it trained on? Tools trained on data similar to your protein of interest will perform better. Ask about the training set composition.
Was it validated on held-out data? Any tool can overfit its training set. Look for performance on proteins the model never saw during training.
Does it report uncertainty? Good models tell you when they're confident and when they're guessing. If a tool gives you a single number with no error bar, be skeptical.
Has it been tested experimentally? Computational benchmarks are necessary but not sufficient. Look for papers where PLM predictions were validated in the lab.
Is it appropriate for your protein? PLMs trained primarily on globular, soluble proteins may perform poorly on membrane proteins, IDPs, or viral proteins. Check whether your protein type was represented in training.
Red Flags
Claims of >90% accuracy without specifying what "accuracy" means
No comparison to simple baselines (e.g., conservation-based methods)
Trained and tested on the same dataset
No error bars or uncertainty estimates
Works "on all proteins" with no caveats
The Bottom Line
Question | Answer |
|---|---|
"What is a protein language model?" | A neural network trained on millions of sequences to learn evolutionary patterns, without manual labels |
"What is ESM-2?" | Meta's 650M-parameter PLM, the most widely used model for protein representation |
"What are embeddings?" | 1,280-number fingerprints per residue that encode everything the model learned about that position |
"What is masked language modeling?" | The training task: hide residues, predict them back. Forces the model to learn sequence patterns. |
"Do PLMs replace AlphaFold?" | No. PLMs give sequence-level representations; AlphaFold gives 3D structures. They're complementary. |
"Should I use PLM-based tools?" | Yes, for variant prioritization and function prediction. No, as a replacement for experimental validation. |
"Are bigger models better?" | Marginally. ESM-2 (650M) is the practical sweet spot. |
Protein language models are the most significant methodological advance in computational protein science since AlphaFold. They won't replace your experiments, but they'll help you design better ones.
References
Rives A, et al. (2021). Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. PNAS, 118(15):e2016239118. Link
Lin Z, et al. (2023). Evolutionary-scale prediction of atomic-level protein structure with a language model. Science, 379(6637):1123-1130. Link
Meier J, et al. (2021). Language models enable zero-shot prediction of the effects of mutations on protein function. NeurIPS, 35. PMC8886682
Dauparas J, et al. (2022). Robust deep learning-based protein sequence design using ProteinMPNN. Science, 378(6615):49-56. Link
Vaswani A, et al. (2017). Attention is all you need. NeurIPS, 30. Link
Rao RM, et al. (2021). MSA Transformer. ICML, 139:8844-8856. Link
Ruff KM & Pappu RV. (2021). AlphaFold and implications for intrinsically disordered proteins. Journal of Molecular Biology, 433(20):167208. Link
Elnaggar A, et al. (2022). ProtTrans: Toward understanding the language of life through self-supervised learning. IEEE Transactions on Pattern Analysis and Machine Intelligence, 44(10):7112-7127. Link
Notin P, et al. (2022). Tranception: protein fitness prediction with autoregressive transformers and inference-time retrieval. ICML. PMC9477541
Hsu C, et al. (2022). Learning inverse folding from millions of predicted structures. ICML. Link
Frazer J, et al. (2021). Disease variant prediction with deep generative models of evolutionary data. Nature, 599:91-95. Link
Book a 20-Minute Demo
Sign up free for unlimited Overview runs — summary, sequence-based analysis, homology search. For the full Characterization — PTMs, binding sites, stability variants, construct design — book a demo and we'll run your target live.
