Blog
Orbion Team
Homology Modeling in the AlphaFold Era: What Still Matters

AlphaFold changed everything. You type in a sequence, wait a few minutes, and get a structure that's often within 1 Å RMSD of the experimental result. So why would anyone still bother with homology modeling? Swiss-Model, MODELLER, I-TASSER—relics of a pre-2021 world, right?
Not quite. AlphaFold solved the structure prediction problem for most single-domain proteins with sufficient homologs. But there are entire categories of problems where homology modeling approaches still matter—and understanding when to trust AlphaFold versus when to reach for other tools is a skill that separates productive structural biologists from those who waste time on overconfident predictions.
Key Takeaways
AlphaFold outperforms homology modeling for nearly all standard single-chain structure predictions—this debate is settled
Homology modeling still matters for: complexes with specific templates, proteins with bound ligands/cofactors, engineered chimeras, and comparative analysis across homologs
Template-based approaches provide context that AlphaFold doesn't: ligand positions, crystal waters, metal coordination, conformational states captured in experimental structures
The real skill is knowing when AlphaFold's confidence metrics signal problems and when template-based methods or experimental structures provide critical information AlphaFold misses
Homology search itself remains essential: finding related structures with known function, ligands, or biology is often more valuable than the model itself
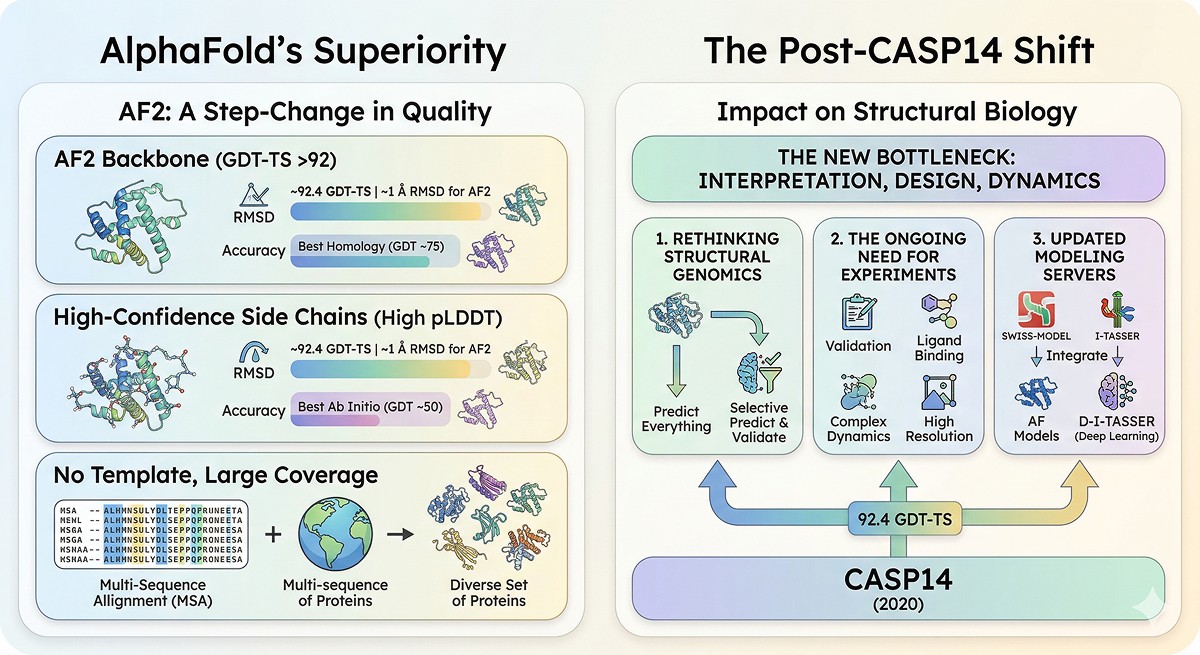
The State of Play: What AlphaFold Changed
What AlphaFold Does Better Than Everything Else
For single-chain protein structure prediction, AlphaFold2 is unambiguously the best method available (Jumper et al., 2021):
Metric | AlphaFold2 | Best Homology Modeling | Best Ab Initio (pre-AF) |
|---|---|---|---|
Median GDT-TS (CASP14) | 92.4 | ~70–80 | ~40–60 |
Backbone accuracy | ~1 Å RMSD for well-predicted regions | ~1–3 Å RMSD | >3 Å RMSD |
Side chain accuracy | Good for high-pLDDT regions | Moderate | Poor |
No template needed | Correct—uses MSA, not explicit templates | Requires template structure | No template needed |
Coverage | Works for most proteins with sufficient MSA depth | Limited to proteins with detectable homologs | Any sequence, but variable quality |
What Changed After CASP14
After AlphaFold's CASP14 results in 2020:
Structural genomics shifted: Many projects pivoted from "predict every structure" to "predict and validate selectively"
PDB submissions continued: Experimental structures remain essential for validation, ligand binding, dynamics, and resolution beyond AlphaFold's capability
Homology modeling servers updated: Swiss-Model now integrates AlphaFold models as templates; I-TASSER evolved into D-I-TASSER with deep learning components
The bottleneck moved: Structure prediction is largely solved; the bottleneck is now interpretation, dynamics, and design
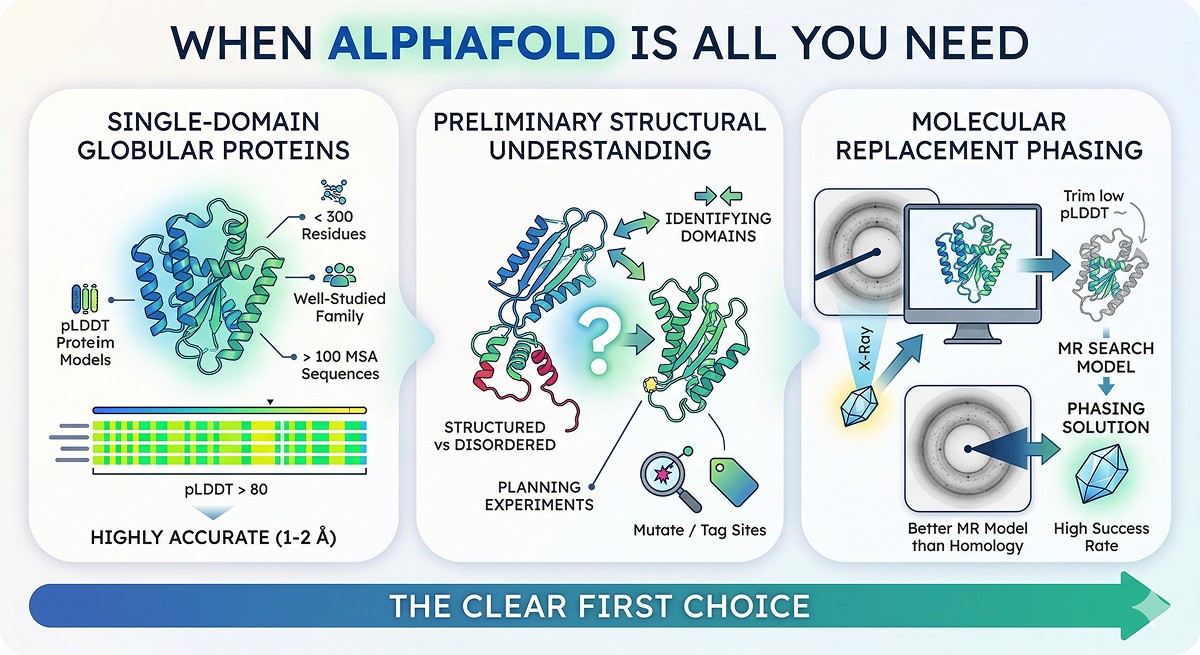
When AlphaFold Is All You Need
For these use cases, run AlphaFold and move on:
Single-Domain Globular Proteins
If your protein is:
A single domain (<300 residues)
From a well-studied protein family
Has >100 sequences in its MSA
pLDDT > 80 across most residues
The AlphaFold model is likely within 1–2 Å of the experimental structure. No homology modeling will improve on it.
Preliminary Structural Understanding
If you need a quick answer to "what does this protein look like?" AlphaFold delivers in minutes with confidence metrics that homology modeling never provided. For:
Planning experiments (which residues to mutate, where to place tags)
Understanding domain architecture
Identifying structured vs disordered regions
AlphaFold is the clear first choice.
Molecular Replacement Phasing
AlphaFold models work as molecular replacement search models for X-ray crystallography with high success rates (McCoy et al., 2022). Trim low-pLDDT regions and you have a better MR model than traditional homology models in most cases.
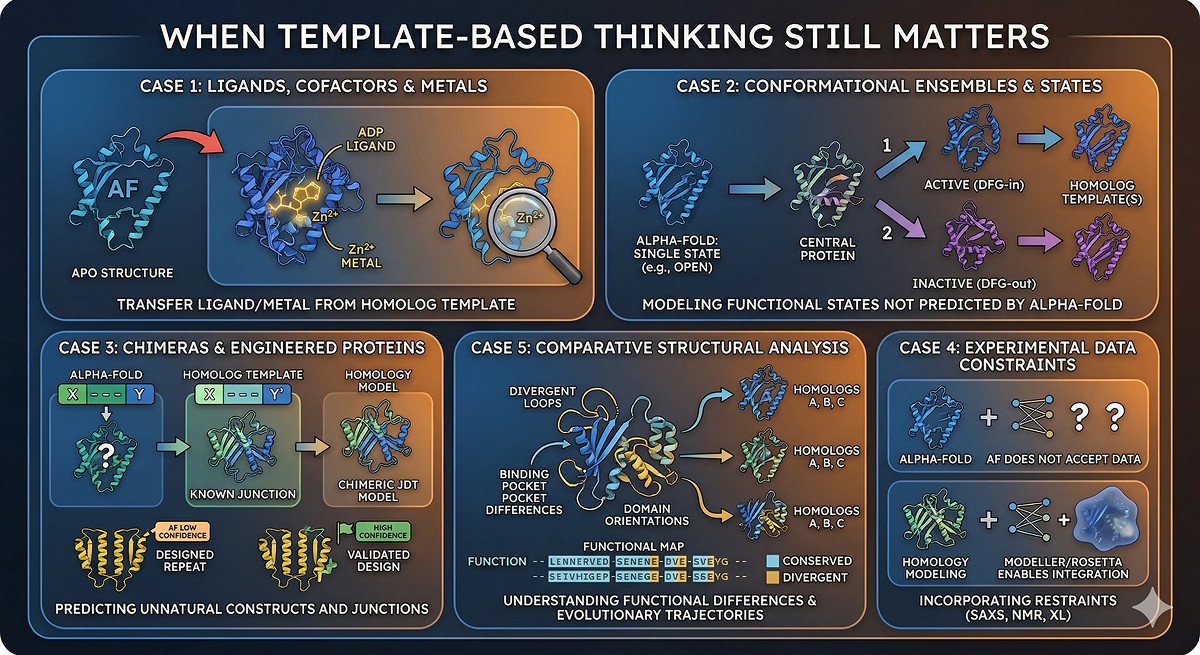
When Homology Modeling (or Template-Based Thinking) Still Matters
Case 1: You Need a Ligand, Cofactor, or Metal in the Model
The problem: AlphaFold predicts the protein structure. It does not predict where ligands, cofactors, metals, or substrates bind. The apo structure may differ significantly from the holo structure.
Why templates help:
If a homologous protein has been co-crystallized with a ligand:
Superpose your AlphaFold model onto the template
Transfer the ligand coordinates
You now have a starting model for docking or structure-based design
Example: You're working on a novel kinase. AlphaFold gives you the apo structure. But PDB has 50 kinase structures with ATP analogs, inhibitors, and substrates bound. Superposing your model onto these templates gives you immediate insight into the binding site geometry with a ligand present.
This is not "homology modeling" in the traditional sense—it's template-based structural annotation. But it uses the same skills and databases.
Case 2: Specific Conformational States
The problem: AlphaFold typically predicts one conformational state. Many proteins exist in multiple states that are functionally important.
Examples:
GPCRs: Active vs inactive conformations (Heo & Bhatt, 2022)
Kinases: DFG-in vs DFG-out
Transporters: Inward-facing vs outward-facing
Enzymes: Open vs closed (substrate-bound vs free)
If an experimental structure of a homolog in the desired state exists, template-based modeling of that state may be more informative than AlphaFold's single-state prediction.
Emerging solution: AlphaFold3 and flow-matching methods are beginning to sample conformational ensembles, but template-based approaches remain more reliable for known functional states.
Case 3: Engineered and Chimeric Proteins
The problem: AlphaFold was trained on natural sequences. It handles them well. But engineered constructs—circular permutants, domain swaps, chimeric proteins, de novo designs—may confuse the model.
When templates are better:
If you've swapped domain A from protein X with domain A' from protein Y, AlphaFold may not know how to handle the junction
If you have an experimental structure of a similar chimera, template-based modeling is more reliable
For designed repeat proteins or computationally designed folds, AlphaFold may assign low confidence even though the design was validated experimentally
Case 4: Modeling with Specific Constraints
The problem: Sometimes you have experimental data (cross-links, NMR restraints, SAXS envelope) that should constrain the model.
Why templates/integrative approaches help:
AlphaFold doesn't accept experimental restraints as input
Homology modeling programs (MODELLER, ROSETTA) can incorporate distance restraints
Integrative modeling frameworks (IMP, HADDOCK) combine templates with experimental data
Case 5: Comparative Structural Analysis
The problem: You want to understand how your protein differs from its homologs—not just what it looks like, but how specific differences affect structure.
Why homology matters:
AlphaFold gives you a model. But understanding why your protein has a different loop conformation, a different binding pocket shape, or a different domain orientation requires comparing it to related structures. This is fundamentally a homology-based analysis:
Find all homologs with experimental structures
Align structures to identify conserved vs divergent regions
Map sequence differences onto structural differences
Generate hypotheses about functional consequences
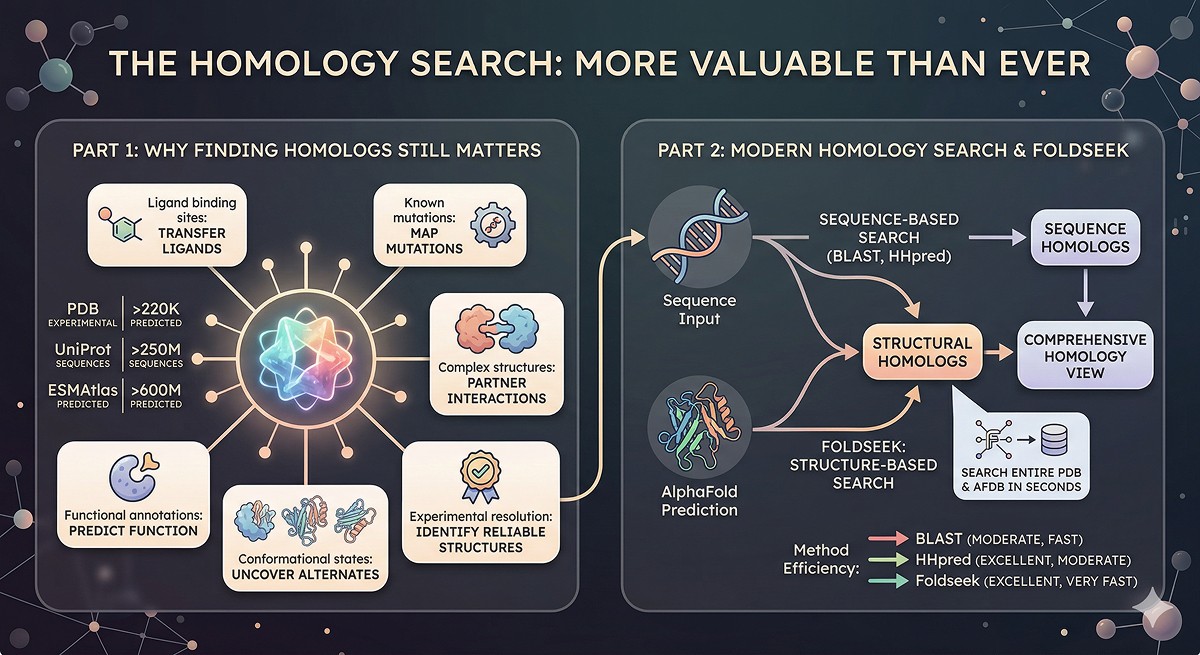
The Homology Search: More Valuable Than Ever
Why Finding Homologs Still Matters
Even if you don't build a homology model, finding structural homologs is essential for interpreting your AlphaFold prediction:
Information from Homologs | How It Helps |
|---|---|
Ligand binding sites | Transfer ligand positions to your model |
Known mutations | Map disease mutations, engineering data from related proteins |
Functional annotations | Enzyme mechanisms, binding partners, pathways |
Conformational states | Access to states AlphaFold doesn't predict |
Experimental resolution | Identify which homolog structures are most reliable |
Complex structures | How homologs interact with partners |
The Scale of Available Data
PDB: >220,000 experimental structures (and growing)
UniProt: >250 million protein sequences
AlphaFold DB: >200 million predicted structures
ESMAtlas: >600 million predicted structures
The challenge is no longer "can I find a structure?" but "which of the 50 available structures is most informative for my question?"
Beyond BLAST: Modern Homology Search
Method | Sensitivity | Speed | Best For |
|---|---|---|---|
BLAST | Moderate (detects >30% identity) | Fast | Quick first-pass search |
PSI-BLAST | Good (iterative profile search) | Moderate | Detecting remote homologs |
HHpred/HHsearch | Excellent (profile-profile comparison) | Moderate | Most sensitive sequence search |
Foldseek | Excellent (structure-based) | Very fast | Finding structural homologs even without sequence similarity |
DALI | Excellent (structure-based) | Slow | Comprehensive structural comparison |
The game-changer: Foldseek enables structural similarity searches at sequence-search speeds. You can now search the entire AlphaFold DB and PDB by structure in seconds—finding homologs that share no detectable sequence similarity but have the same fold.
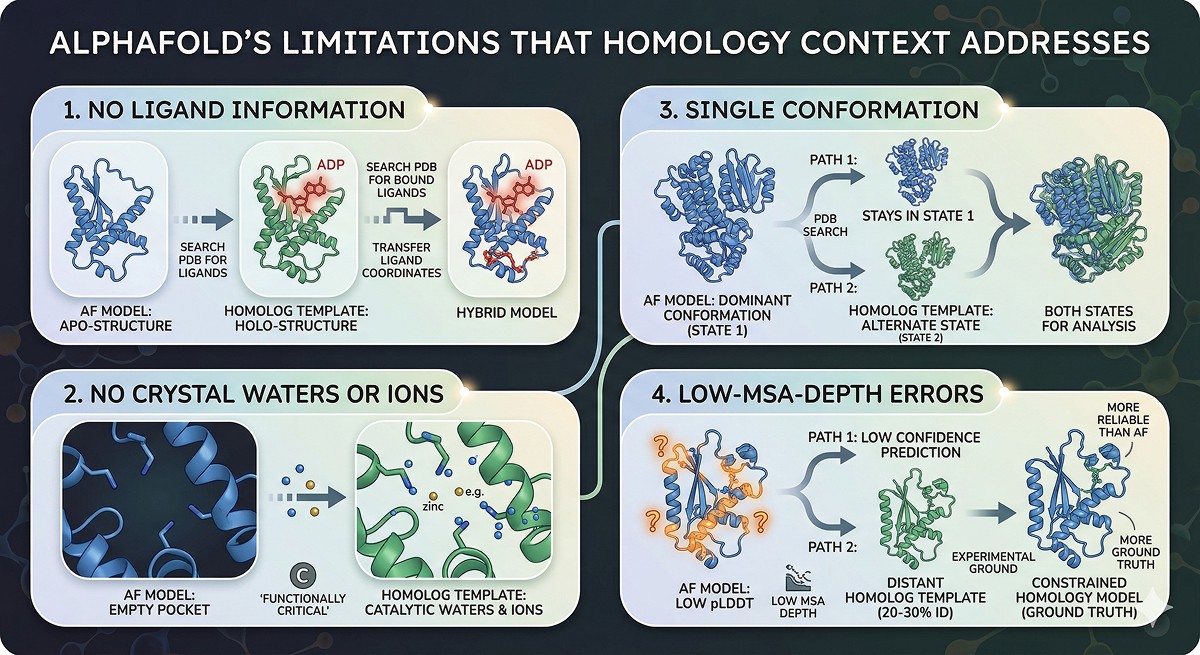
AlphaFold's Limitations That Homology Context Addresses
Limitation 1: No Ligand Information
AlphaFold models are apo structures. For drug discovery, you need holo structures:
Workflow:
Run AlphaFold → get the fold
Search PDB for homologs with bound ligands (Foldseek or BLAST)
Superpose → transfer ligand coordinates
Use as starting point for docking or MD
This hybrid workflow gives you the best of both worlds: AlphaFold's accurate backbone + template-derived ligand context.
Limitation 2: No Crystal Waters or Ions
Water molecules and ions are often functionally critical:
Catalytic waters in enzyme active sites
Structural zinc ions
Bridging waters at protein-protein interfaces
These are present in experimental structures of homologs. Transferring them to your AlphaFold model improves the biological relevance of the model.
Limitation 3: Single Conformation
AlphaFold predicts the "average" or "dominant" conformation. For proteins that switch states:
Workflow:
AlphaFold → state 1 (probably the dominant conformation)
PDB search → find homologs in state 2
Model state 2 by homology or by using AlphaFold with MSA subsampling
Now you have both states for analysis
Limitation 4: Prediction Errors in Low-MSA-Depth Proteins
For proteins from underrepresented organisms or novel folds, AlphaFold's predictions may be unreliable (low pLDDT). In these cases:
If a distant homolog (20–30% identity) has an experimental structure, a constrained homology model may be more reliable than a low-confidence AlphaFold prediction
The experimental template provides "ground truth" for at least some structural features
This is increasingly rare as AlphaFold coverage expands, but still relevant for viral proteins, extremophile proteins, and recently diverged families
The Modern Structural Biology Workflow
The Integrated Approach
The best current practice combines AlphaFold with homology-based context:
When to Run What
Situation | Best Approach | Why |
|---|---|---|
Need structure of a single protein | AlphaFold | Best accuracy, fastest |
Need structure with ligand bound | AlphaFold + template transfer | AF doesn't predict ligands |
Need specific conformational state | Template from homolog in that state | AF predicts one state |
Working with designed/chimeric protein | Template if available; AF as supplement | AF may not handle non-natural junctions |
Low-MSA-depth protein | Both; compare and use experimental template if available | AF may be unreliable without MSA depth |
Need to understand functional differences across a family | Structural alignment of AF models + experimental structures | Comparative analysis |
Crystal structure phasing (MR) | AlphaFold model, trimmed to high-pLDDT regions | Superior MR search model |
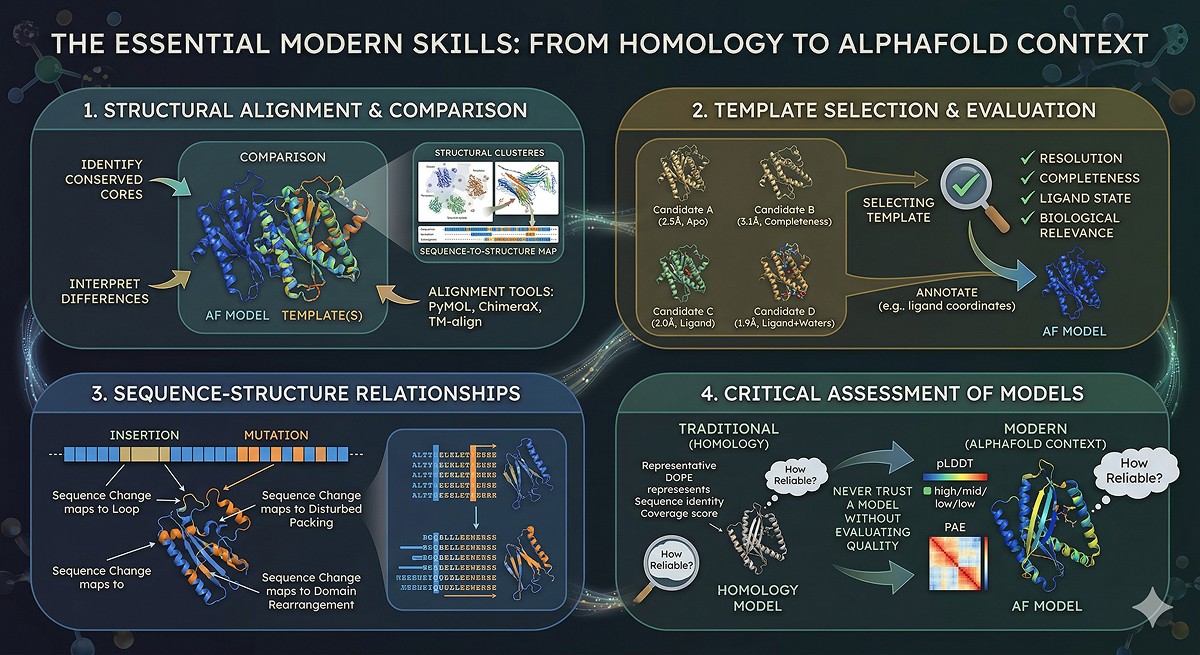
The Skills That Transferred
Even though homology modeling as a standalone method is largely obsolete, the skills it required are more valuable than ever:
Structural Alignment and Comparison
Understanding how to align structures, identify conserved cores, and interpret structural differences is essential for using AlphaFold models in context. Tools: PyMOL align/super, ChimeraX matchmaker, TM-align.
Template Selection and Evaluation
Knowing which experimental structure is most informative for your question—considering resolution, completeness, ligand state, and biological relevance—transfers directly to the modern workflow of annotating AlphaFold models with homolog data.
Sequence-Structure Relationships
Understanding how sequence changes map to structural changes—insertions creating loops, mutations disrupting packing, domain rearrangements—is fundamental to interpreting any structural model, whether from AlphaFold or experiment.
Critical Assessment of Models
The habit of asking "how reliable is this model?" and using quantitative metrics to answer that question is more important than ever. AlphaFold provides pLDDT and PAE. Homology modeling provided sequence identity, coverage, and DOPE scores. The skill is the same: never trust a model without evaluating its quality.
The Bottom Line
Question | AlphaFold | Homology / Template-Based | Both |
|---|---|---|---|
"What does my protein look like?" | Best choice | Obsolete for this | — |
"Where does the ligand bind?" | Can't answer this | Transfer from homolog | Use together |
"What conformational states exist?" | Predicts one state | Access to other states via templates | Use together |
"How does my protein compare to homologs?" | Provides a model | Provides context | Essential combination |
"Can I trust this model region?" | pLDDT + PAE | Experimental template as validation | Cross-reference |
"What function does this protein have?" | No direct prediction | Functional annotation from homologs | Homology search is key |
The core message: AlphaFold made structure prediction routine. It did not make structural biology routine. Understanding proteins still requires the comparative, contextual thinking that was always at the heart of homology-based approaches—it just starts from a much better structural model now.
Structural Context and Homology at Scale with Orbion
Orbion integrates AlphaFold2 structure prediction with homology search across over 700 million proteins from UniProt and PDB—all within Orbion's own infrastructure, so sequences are never sent to third-party services. For every submitted protein, the platform generates a structural model with full pLDDT and PAE confidence metrics, then automatically searches for homologs to provide functional context: what's known about related proteins, their annotations, and their experimental characterization.
The PAE Insight Engine extracts domain boundaries, hinge regions, and conformational heterogeneity cues from the confidence data—the kind of interpretation that used to require expert manual analysis of homology models. Combined with AstraROLE for functional annotation and AstraBIND for binding site prediction, researchers get the structural model AND the biological context that makes it interpretable—bridging the gap between "here's the structure" and "here's what it means."
References
Jumper J, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596:583-589. Link
McCoy AJ, et al. (2022). Implications of AlphaFold2 for crystallographic phasing by molecular replacement. Acta Crystallographica Section D, 78(1):1-13. PMC8662965
Heo L & Bhatt S. (2022). Assessment of AlphaFold predictions of GPCR structures. bioRxiv. PMC9348835
van Kempen M, et al. (2024). Fast and accurate protein structure search with Foldseek. Nature Biotechnology, 42:243-246. Link
Waterhouse A, et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Research, 46(W1):W296-W303. PMC6030848
Webb B & Sali A. (2016). Comparative protein structure modeling using MODELLER. Current Protocols in Bioinformatics, 54:5.6.1-5.6.37. PMC5031415
Akdel M, et al. (2022). A structural biology community assessment of AlphaFold2 applications. Nature Structural & Molecular Biology, 29:1056-1067. Link
Varadi M, et al. (2022). AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space. Nucleic Acids Research, 50(D1):D439-D444. PMC8728224
Zimmermann L, et al. (2018). A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. Journal of Molecular Biology, 430(15):2237-2243. Link
Terwilliger TC, et al. (2024). AlphaFold predictions are useful for nearly all proteins in the PDB. Nature, 634:931-935. Link
Book a 20-Minute Demo
Sign up free for unlimited Overview runs — summary, sequence-based analysis, homology search. For the full Characterization — PTMs, binding sites, stability variants, construct design — book a demo and we'll run your target live.
