Blog
Orbion Team
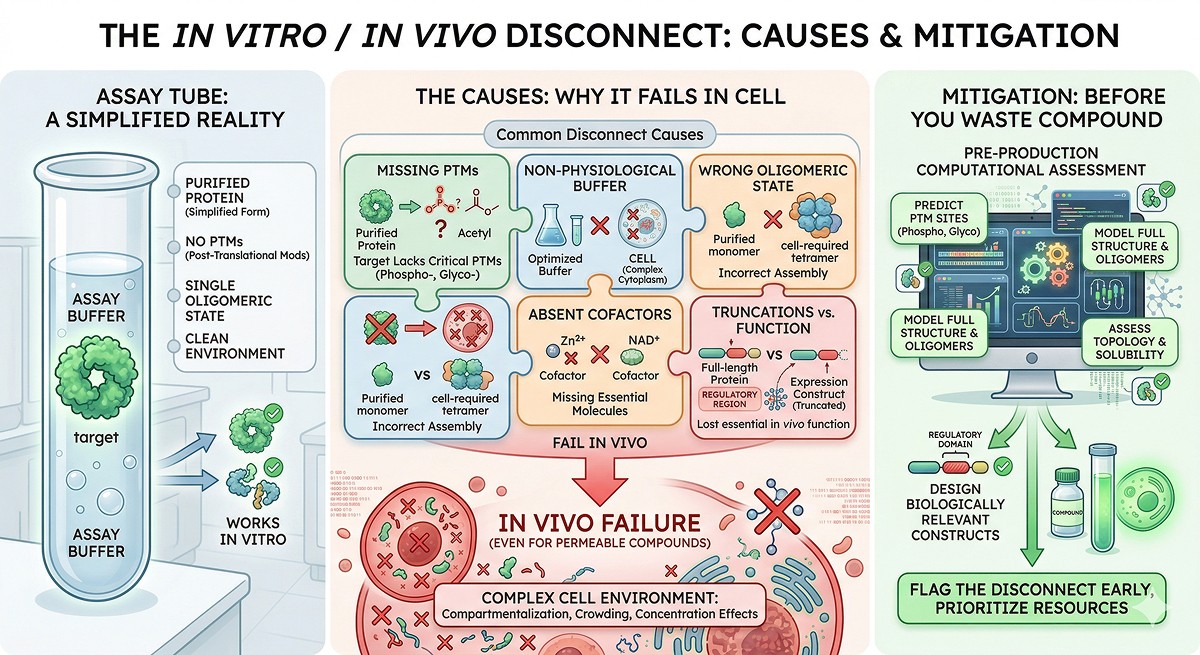
Why Your Protein Works in the Assay but Fails in Cells

Your enzyme inhibitor has an IC50 of 12 nM in the biochemical assay. Clean dose-response. Competitive mechanism confirmed. You move to cells, expecting a potent hit. The EC50 in the cell-based assay? Greater than 10 µM. An 800-fold drop in potency. Your medicinal chemistry team is skeptical. Your biologist says "the compound doesn't work." But the compound works perfectly—against a purified, recombinant, truncated protein in an optimized buffer that looks nothing like the inside of a cell.
The in vitro-to-in vivo translation gap is one of the most expensive problems in drug discovery and protein science. Understanding why it happens—and how to predict it—saves months of failed cell-based experiments.
Key Takeaways
Purified recombinant proteins are simplified versions of reality: they lack post-translational modifications, binding partners, compartmentalization, and cellular concentrations
The most common causes of in vitro/in vivo disconnect: missing PTMs, wrong oligomeric state, absent cofactors, non-physiological buffer conditions, and missing protein-protein interactions
Cell permeability is not the only explanation: even membrane-permeable compounds fail when the target protein behaves differently in cells
Construct design choices propagate to biological relevance: truncations that improve expression can remove regulatory regions essential for in vivo function
Computational assessment of PTMs, topology, and suitability before protein production can flag the disconnect before you waste compound
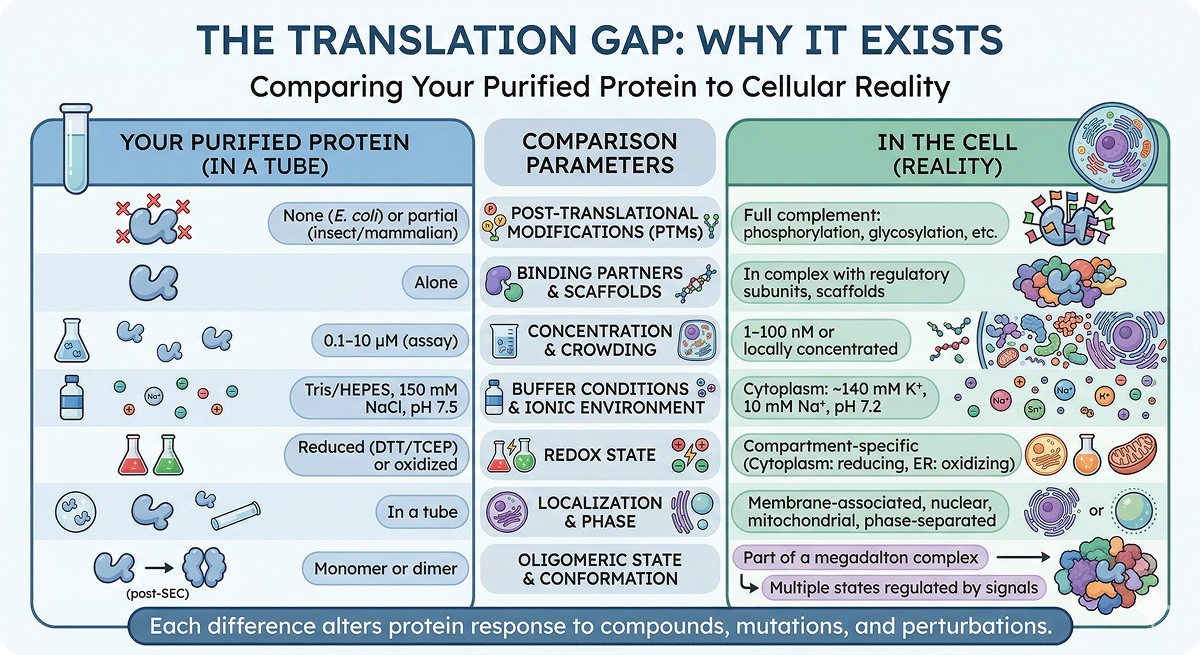
The Translation Gap: Why It Exists
What Your Purified Protein Is Missing
When you express a protein in E. coli, purify it, and put it in an assay, you've created a version of the protein that differs from the cellular reality in multiple ways:
Property | Your Purified Protein | In the Cell |
|---|---|---|
PTMs | None (E. coli) or partial (insect/mammalian) | Full complement: phosphorylation, glycosylation, ubiquitination, acetylation |
Binding partners | Alone | In complex with regulatory subunits, scaffolds, substrates |
Concentration | 0.1–10 µM (assay) | 1–100 nM (typical cellular) or locally concentrated in compartments |
Buffer | Tris/HEPES, 150 mM NaCl, pH 7.5 | Cytoplasm: ~140 mM K⁺, 10 mM Na⁺, pH 7.2, crowded with macromolecules |
Redox state | Reduced (DTT/TCEP) or oxidized (no reducing agent) | Compartment-specific (cytoplasm: reducing; ER: oxidizing) |
Localization | In a tube | Membrane-associated, nuclear, mitochondrial, or phase-separated |
Oligomeric state | Monomer or dimer (post-SEC) | May be part of a megadalton complex |
Conformation | One state (often apo) | Multiple states regulated by signals |
Every one of these differences can change how your protein responds to a compound, mutation, or experimental perturbation.
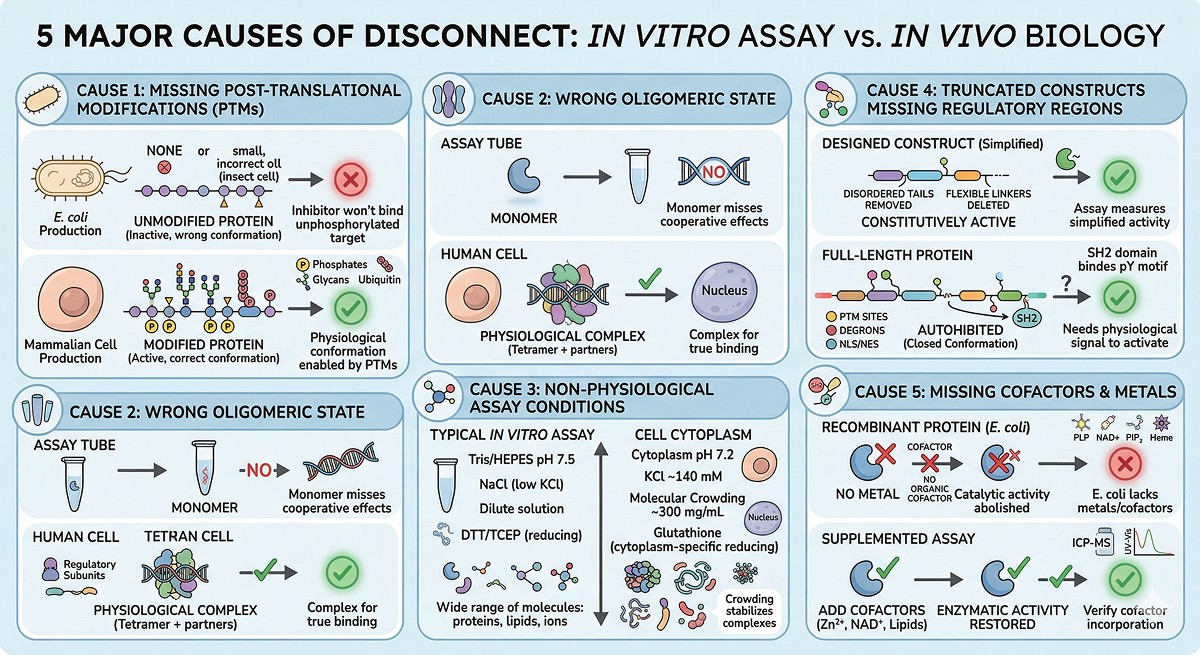
The Five Major Causes of Disconnect
Cause 1: Missing Post-Translational Modifications
This is the most common and most underappreciated cause.
Phosphorylation:
~75% of human proteins are phosphorylated at one or more sites
Phosphorylation can activate, inhibit, or alter substrate specificity
Your E. coli-expressed kinase has no regulatory phosphorylation → it may be constitutively active or inactive
An inhibitor that binds the phosphorylated form won't bind your unphosphorylated recombinant protein (or vice versa)
Example: EGFR
EGFR autophosphorylation at Y1068, Y1086, and other sites creates docking sites for downstream signaling proteins. The unphosphorylated form has a different conformation and different binding properties. Assays using unphosphorylated EGFR may identify compounds that don't engage the physiologically relevant form.
Glycosylation:
Glycans affect folding, stability, receptor binding, and drug access
E. coli cannot glycosylate; insect cells produce different glycans than mammalian cells
A binding site partially occluded by a glycan in vivo may be wide open in your recombinant protein
Ubiquitination and degradation:
In cells, your target protein may have a half-life of 30 minutes
Stabilizing the target (by inhibiting its degradation pathway) may be more effective than inhibiting its activity
Your purified protein has no degradation pathway—it's artificially stable
Cause 2: Wrong Oligomeric State
The problem: Many proteins function as part of multi-subunit complexes. Purifying a single subunit gives you something that doesn't exist in biology.
Examples:
Protein | In Your Tube | In the Cell | Consequence |
|---|---|---|---|
p53 | Monomer or tetramer | Tetramer bound to DNA, MDM2, and other regulators | Monomer assays miss cooperative effects |
Proteasome | 20S core particle | 26S (20S + 19S regulatory particle) | Inhibitor access differs with regulatory cap |
RNA Pol II | Rpb1 subunit alone | 12-subunit complex + mediator + GTFs | Individual subunit assays are biologically meaningless |
Ion channels | Purified subunit | Tetrameric channel in membrane with auxiliary subunits | Function requires assembly |
The fix: Whenever possible, assay the biologically relevant complex, not an isolated subunit. Co-expression, co-purification, or reconstitution from purified components gives a more physiological target.
Cause 3: Non-Physiological Assay Conditions
Buffer composition matters more than most people realize:
Assay Parameter | Typical In Vitro | Physiological | Effect on Results |
|---|---|---|---|
pH | 7.5 (Tris or HEPES) | 7.2 (cytoplasm), 4.5–6.5 (endosomes/lysosomes) | pH-sensitive binding interactions change |
K⁺ concentration | 0–150 mM NaCl | ~140 mM KCl | Ion selectivity affects metalloenzymes |
Molecular crowding | Dilute solution | ~300 mg/mL macromolecules | Crowding affects binding constants, folding |
Reducing agent | 1 mM DTT or TCEP | Glutathione (1–10 mM cytoplasm) | Affects cysteine-dependent interactions |
ATP | 1 mM (if added) | 1–5 mM (cytoplasm) | ATP-competitive inhibitors face different competition |
Substrate concentration | Usually above Km | Often below Km | Changes apparent inhibitor potency |
Molecular crowding is particularly important: In dilute buffer, weak interactions don't hold. In the crowded cytoplasm (300 mg/mL total protein), the effective concentration of binding partners is much higher, and excluded volume effects stabilize complexes that fall apart in your assay (Ellis & Minton, 2003).
Cause 4: Truncated Constructs Missing Regulatory Regions
The construct design problem:
To get your protein to express and purify, you probably:
Removed disordered N- and C-terminal tails
Deleted flexible linkers between domains
Truncated to a single domain for crystallization
These removed regions often contain:
Regulatory phosphorylation sites (frequently in disordered loops/tails)
Degron sequences (control protein half-life)
Autoinhibitory domains (keep the enzyme inactive until activated by a signal)
Protein-protein interaction motifs (SH2-binding pYXXφ motifs, SH3-binding PxxP motifs)
Localization signals (NLS, NES, membrane anchors)
Example: Kinase autoinhibition
Many kinases have autoinhibitory segments outside the catalytic domain. The truncated kinase domain is constitutively active. An inhibitor that binds the autoinhibited (closed) conformation won't work on the constitutively active truncation—and vice versa.
Example: Nuclear localization
If your protein has a nuclear localization signal in the truncated region, your assay is measuring cytoplasmic behavior of a protein that only functions in the nucleus.
Cause 5: Missing Cofactors and Metals
Many proteins require cofactors that aren't present in your assay:
Cofactor Type | Examples | Consequence When Missing |
|---|---|---|
Metal ions | Zn²⁺, Mg²⁺, Fe²⁺/³⁺, Ca²⁺, Mn²⁺ | Catalytic activity abolished or altered |
Organic cofactors | NAD⁺/NADH, FAD, PLP, CoA, SAM | No enzymatic activity |
Lipids | PIP2, cholesterol, specific phospholipids | Membrane proteins: altered conformation |
Nucleotides | GTP (G-proteins), ATP (kinases) | Wrong activation state |
Heme | Iron porphyrin | Cytochrome P450s: no function without heme |
The E. coli problem: Recombinant proteins from E. coli often lack the correct metal or cofactor:
Zinc metalloproteases may incorporate nickel from Ni-NTA purification
Iron-sulfur cluster proteins lose their clusters during aerobic purification
PLP-dependent enzymes may purify without PLP if it wasn't supplemented
The fix: Check what cofactors your protein needs before designing assays. Add them to the assay buffer. Better yet, verify incorporation by UV-Vis, ICP-MS (for metals), or activity assays with/without cofactor supplementation.
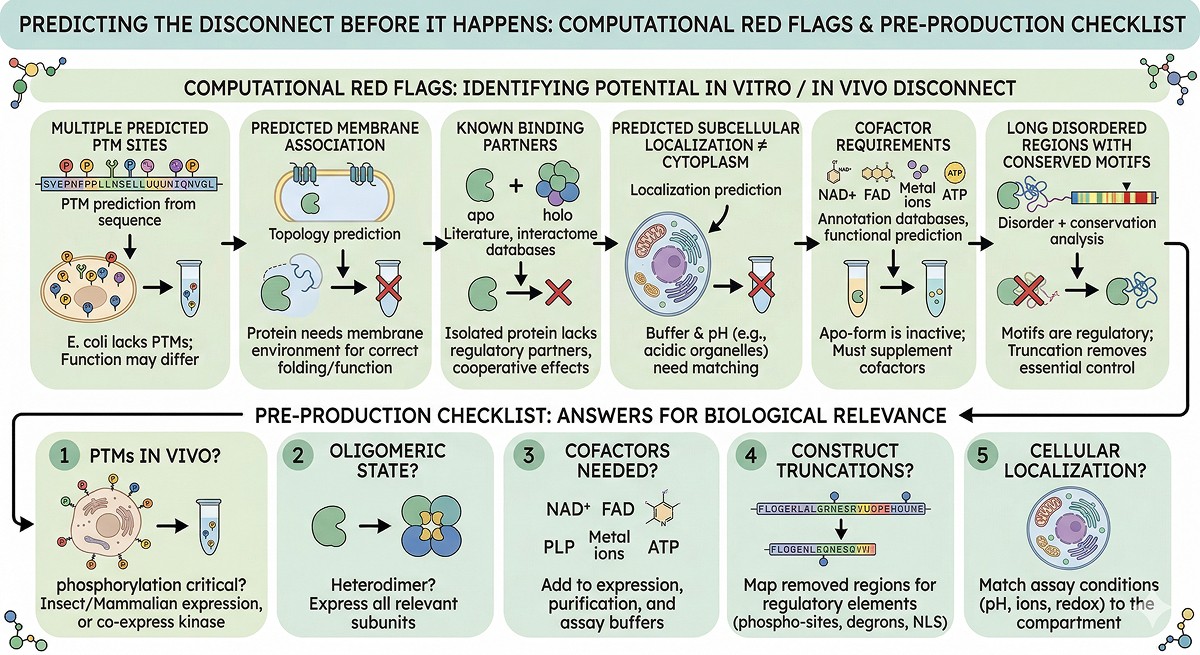
Predicting the Disconnect Before It Happens
Computational Red Flags
Before you even start producing protein, you can flag potential in vitro/in vivo disconnect:
Red Flag | How to Check | Implication |
|---|---|---|
Multiple predicted PTM sites | PTM prediction from sequence | E. coli protein will lack these; function may differ |
Predicted membrane association | Topology prediction | Protein may need a membrane environment |
Known binding partners | Literature, interactome databases | Isolated protein may behave differently |
Predicted subcellular localization ≠ cytoplasm | Localization prediction | Buffer and pH may need adjustment |
Cofactor requirements | Annotation databases, functional prediction | Must supplement in assay |
Long disordered regions with conserved motifs | Disorder + conservation analysis | These motifs may be regulatory; truncation removes regulation |
The Pre-Production Checklist
Before expressing your protein, answer these questions:
What PTMs does it have in vivo? If phosphorylation is critical, consider insect or mammalian expression, or co-express with the relevant kinase.
What oligomeric state is biologically relevant? If it's a heterodimer, express both subunits.
What cofactors does it need? Add them to expression, purification, and assay buffers.
What was truncated from your construct? Map the removed regions for regulatory elements.
Where is this protein in the cell? Match your assay conditions to the relevant compartment (pH, ions, redox).
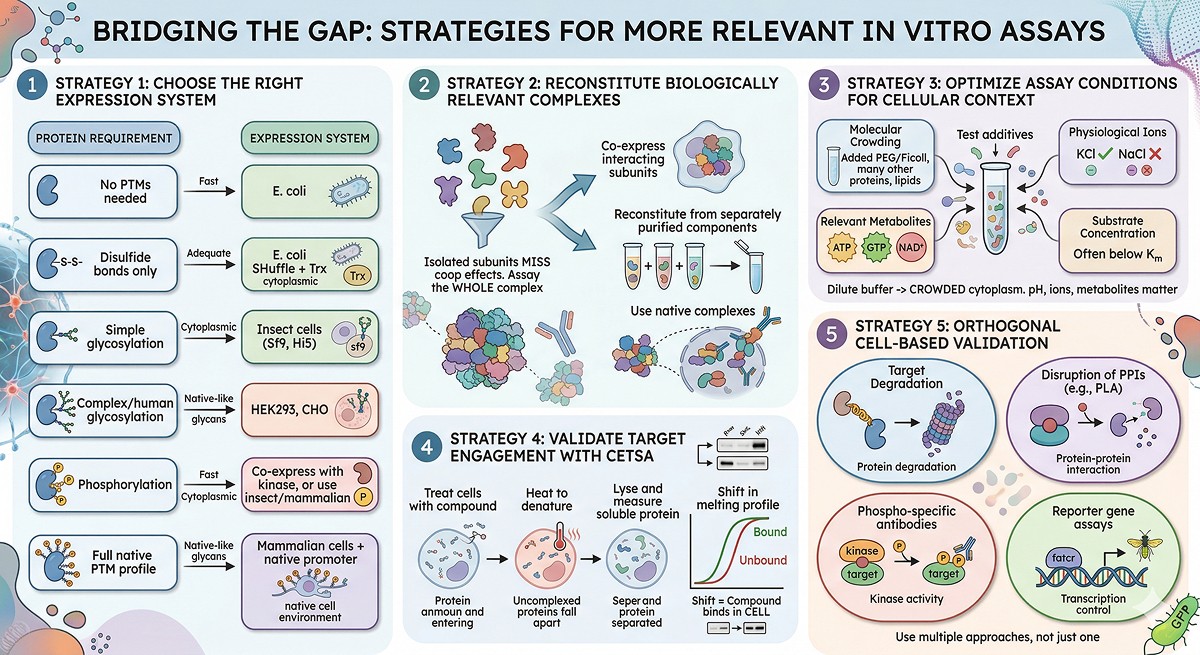
Bridging the Gap: Strategies for More Relevant In Vitro Assays
Strategy 1: Use the Right Expression System
Protein Requirement | Expression System | Why |
|---|---|---|
No PTMs needed | E. coli | Fast, cheap, adequate |
Disulfide bonds only | E. coli SHuffle + Trx | Cytoplasmic disulfides |
Simple glycosylation | Insect cells (Sf9, Hi5) | Core glycans, some processing |
Complex/human glycosylation | HEK293, CHO | Native-like glycan profiles |
Phosphorylation | Co-express with kinase, or use insect/mammalian | Active kinase in host cell |
Full native PTM profile | Mammalian cells + native promoter | Closest to endogenous |
Strategy 2: Reconstitute Complexes
Rather than assaying an isolated subunit:
Co-express interacting subunits
Reconstitute from separately purified components
Use native complexes immunoprecipitated from cells (less pure, but more relevant)
Strategy 3: Modify Assay Conditions
Add molecular crowding agents (PEG, Ficoll) to simulate cytoplasmic crowding
Use physiological ion concentrations (KCl, not NaCl)
Include relevant metabolites (ATP, GTP, NAD⁺)
Test at physiological substrate concentrations (often below Km)
Strategy 4: Validate with Cellular Thermal Shift (CETSA)
CETSA (Cellular Thermal Shift Assay) measures target engagement in intact cells:
Treat cells with compound
Heat to denature unbound protein
Lyse and measure remaining soluble protein by Western blot
Shift in melting profile = compound binds the target in the cellular context
This directly addresses the in vitro/in vivo gap by measuring binding in the native cellular environment.
Strategy 5: Orthogonal Cell-Based Validation
Don't rely on a single cell-based readout. Use multiple approaches:
Target degradation (if mechanism is relevant)
Proximity ligation assay (PLA) for disrupting protein-protein interactions
Phospho-specific antibodies for kinase targets
Reporter gene assays for transcription factor targets
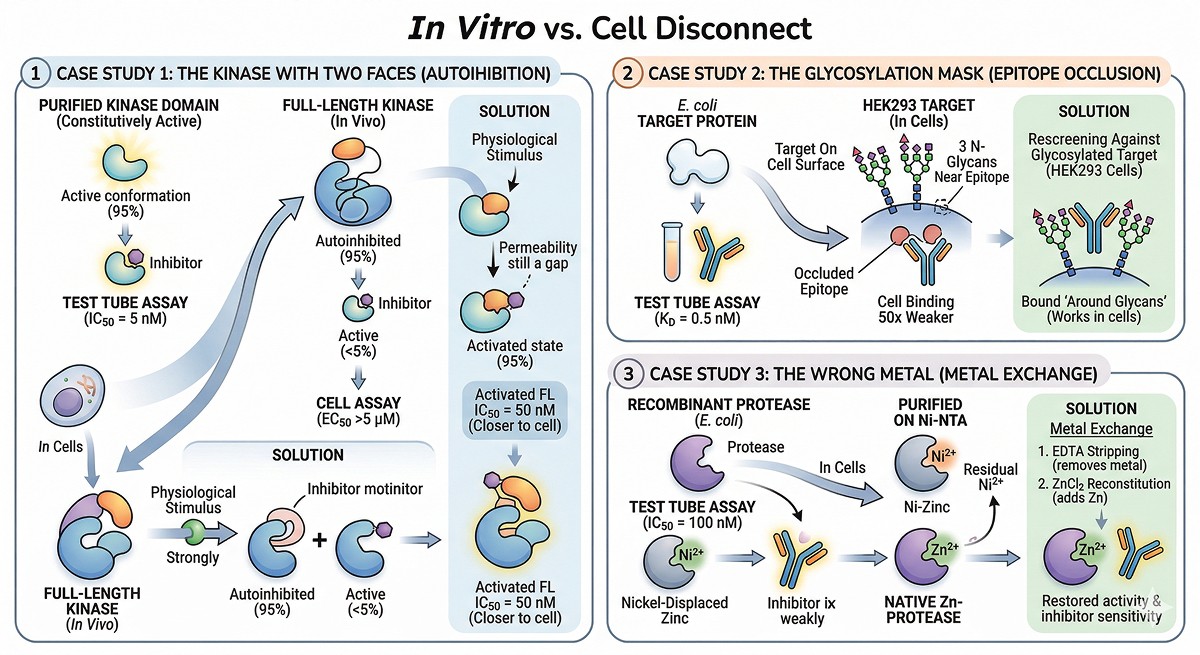
Case Studies: The Disconnect in Action
Case Study 1: The Kinase With Two Faces
A kinase inhibitor had IC50 = 5 nM against the purified kinase domain. In cells, the EC50 was >5 µM.
Root cause: The purified kinase domain was constitutively active (no autoinhibitory domain). In cells, the kinase was 95% autoinhibited. The inhibitor bound the active conformation that represented <5% of the cellular population.
Solution: Expressing the full-length kinase (including the autoinhibitory domain) and activating it with the physiological stimulus before adding inhibitor gave an IC50 of 50 nM—much closer to the cellular EC50 of 200 nM. The remaining gap was cell permeability.
Case Study 2: The Glycosylation Mask
A therapeutic antibody bound its target with KD = 0.5 nM using an E. coli-expressed target protein. Against the same target on cell surfaces, binding was 50-fold weaker.
Root cause: The target protein had three N-glycosylation sites near the antibody epitope. The glycans partially occluded the binding site. The E. coli protein lacked these glycans, presenting a fully exposed epitope that doesn't exist in vivo.
Solution: Re-screening against the glycosylated target (expressed in HEK293 cells) identified antibodies that bound between or around the glycans—antibodies that worked on real cells.
Case Study 3: The Wrong Metal
A metalloprotease inhibitor showed IC50 = 100 nM in the biochemical assay. In cells: no activity at any concentration tested.
Root cause: The recombinant protease was purified on Ni-NTA. Residual nickel displaced the catalytic zinc. The inhibitor was a zinc chelator—with no zinc to chelate, it couldn't work. But in cells, the protease had its native zinc and was in a different conformation.
Solution: Metal exchange (EDTA stripping → ZnCl₂ reconstitution) restored native activity and inhibitor sensitivity.
The Bottom Line
In Vitro Observation | Possible Cellular Reality | How to Check |
|---|---|---|
IC50 = 10 nM | EC50 = 1 µM (autoinhibition in cells) | Test full-length protein, activate physiologically |
Strong binding (KD = 1 nM) | Weak binding in cells (glycan occlusion) | Test against glycosylated target |
Protein is constitutively active | Protein is tightly regulated in cells | Express with regulatory domains/subunits |
Monomeric in SEC | Heterotetrameric in cells | Co-express subunits, check by native PAGE |
Active without cofactor | Requires cofactor for native conformation | Add physiological cofactors |
Soluble in assay buffer | Membrane-anchored in cells | Include lipids or membrane mimics |
The fundamental insight: Your purified protein is a simplified model of reality. Every simplification—truncation, expression system, buffer, concentration—introduces a potential disconnect. The goal isn't to eliminate all simplifications (impossible), but to understand which ones matter for your specific question.
Anticipating the Disconnect with Orbion
Orbion helps flag potential in vitro/in vivo disconnects before protein production begins. AstraPTM predicts 39 post-translational modification types at residue resolution, revealing which sites may be functionally important and which expression system is needed to capture them. AstraSUIT predicts subcellular localization, membrane association, host organism compatibility, and cofactor requirements—each a potential source of disconnect if ignored.
Combined with the AI-generated literature scan that summarizes known biology for each protein, researchers can identify regulatory interactions, binding partners, and functional PTMs from the start. The goal is to make informed decisions about construct design and expression system before cloning—so the protein you produce in the lab resembles the protein that functions in the cell.
References
Ellis RJ & Minton AP. (2003). Cell biology: join the crowd. Nature, 425:27-28. Link
Martinez Molina D, et al. (2013). Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science, 341(6141):84-87. Link
Apweiler R, et al. (1999). On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochimica et Biophysica Acta, 1473(1):4-8. Link
Cohen P. (2000). The regulation of protein function by multisite phosphorylation—a 25 year update. Trends in Biochemical Sciences, 25(12):596-601. PMC493375
Huse M & Bhatt S. (2002). The conformational plasticity of protein kinases. Cell, 109(3):275-282. Link
Scheck A & Bhatt S. (2020). The role of post-translational modifications in protein structure and function. Current Opinion in Structural Biology, 62:67-73.
Raman EP, et al. (2009). Origins of biomolecular force field performance: implications for coarse-grained and multiscale models. Journal of Chemical Theory and Computation, 5(11):3034-3044.
Arkin MR, et al. (2014). Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chemistry & Biology, 21(9):1102-1114. PMC4199827
Huber KVM, et al. (2015). Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature, 508:222-227.
Jafari R, et al. (2014). The cellular thermal shift assay for evaluating drug target interactions in cells. Nature Protocols, 9:2100-2122. Link
Book a 20-Minute Demo
Sign up free for unlimited Overview runs — summary, sequence-based analysis, homology search. For the full Characterization — PTMs, binding sites, stability variants, construct design — book a demo and we'll run your target live.
