Blog
Orbion Team
MBP vs SUMO vs GST vs Thioredoxin: Choosing the Right Fusion Partner

Your protein doesn't express solubly in E. coli. Your advisor says "try MBP." Your labmate swears by SUMO. The postdoc down the hall uses GST for everything. The vendor catalog lists fifteen different fusion systems, each claiming superior results. You have budget for three constructs. If you pick wrong, you've wasted a month of cloning and expression trials. If you pick right, you might go from zero soluble protein to milligrams in a week.
Fusion partner selection is one of the most consequential early decisions in a protein production campaign, yet most researchers choose based on lab tradition rather than rational analysis.
Key Takeaways
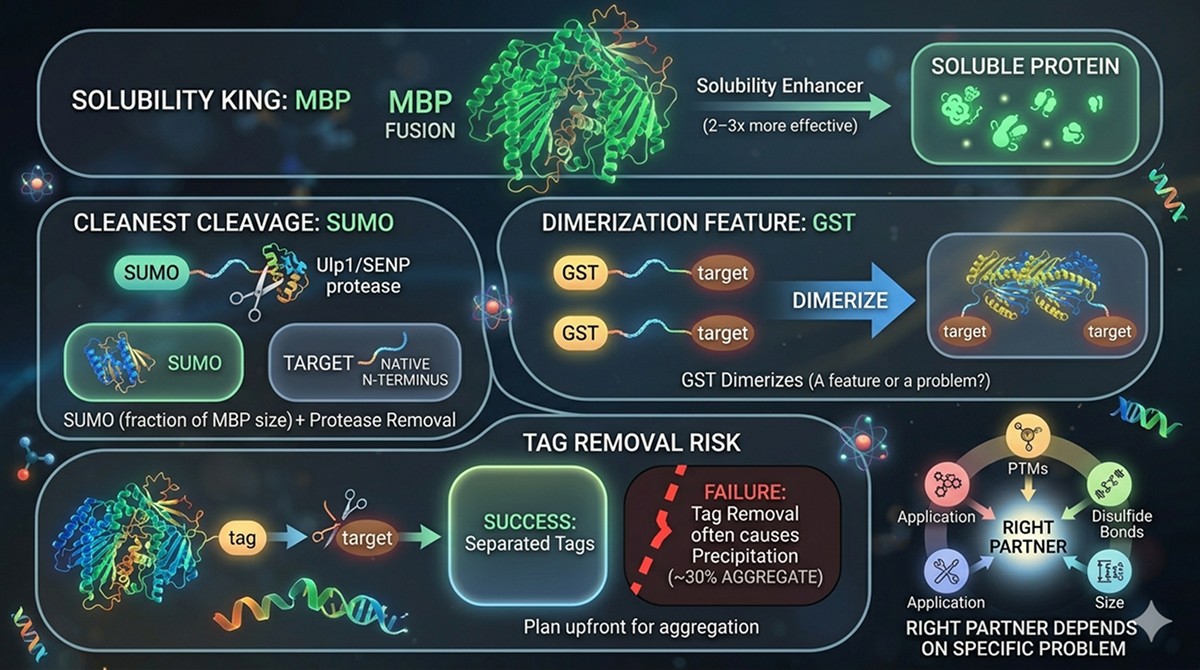
MBP is the most effective general-purpose solubility enhancer, improving soluble expression 2–3x more often than other tags in systematic comparisons
SUMO offers the cleanest cleavage (native N-terminus after Ulp1/SENP removal) with meaningful solubility enhancement at a fraction of MBP's size
GST dimerizes—a feature or a problem depending on your downstream application
Tag removal often causes precipitation: ~30% of proteins that are soluble as fusions aggregate after cleavage, so plan for this upfront
The "right" fusion partner depends on your protein's specific problems: PTM requirements, disulfide bonds, size, and downstream application should drive the choice
Why Fusion Partners Work
The Problem They Solve
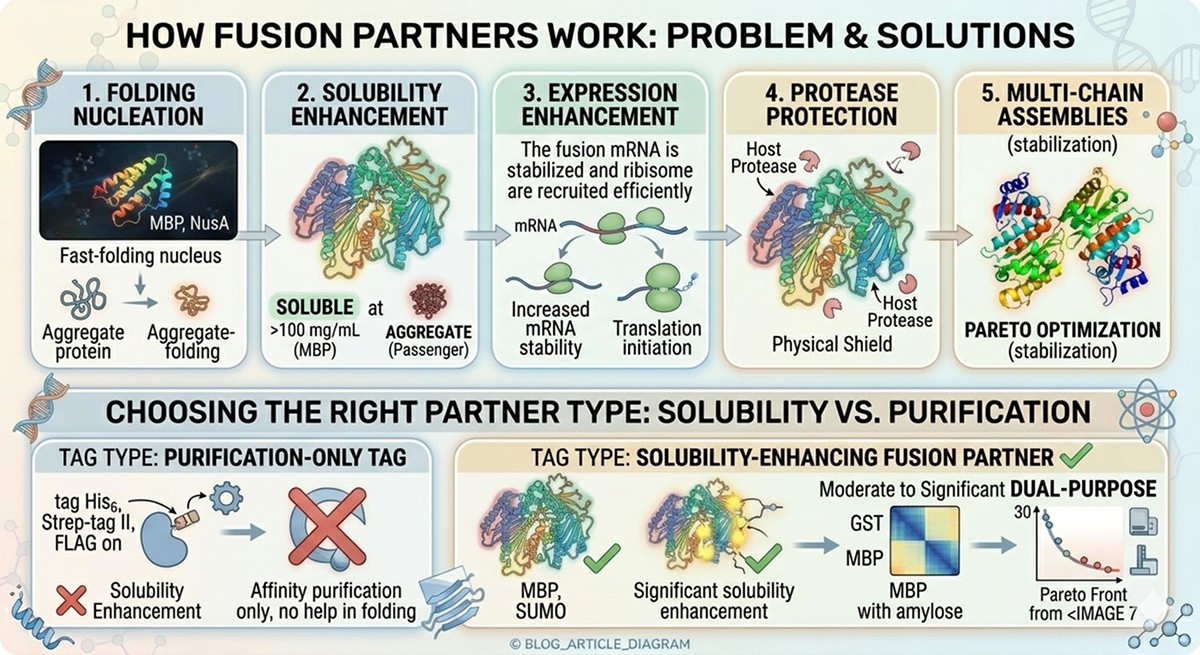
Many recombinant proteins fail to fold properly in E. coli. They aggregate into inclusion bodies, get degraded by host proteases, or express at levels too low to detect. Fusion partners address these problems through several mechanisms (Esposito & Chatterjee, 2006):
Folding nucleation: Large, fast-folding fusion partners (MBP, NusA) fold first and create a local environment that promotes downstream folding of the passenger protein.
Solubility enhancement: Highly soluble fusion partners (MBP is soluble at >100 mg/mL) can keep aggregation-prone passengers in solution during folding.
Expression enhancement: Fusion to a well-expressed protein can increase mRNA stability and translation initiation efficiency.
Protease protection: Large fusion partners can physically shield the passenger protein from host proteases during early expression.
Not All Tags Enhance Solubility
It's critical to distinguish between purification tags and solubility-enhancing fusion partners:
Tag Type | Purpose | Examples | Solubility Enhancement |
|---|---|---|---|
Purification tag | Affinity purification only | His6, Strep-tag II, FLAG | None or minimal |
Solubility enhancer | Improve folding and solubility | MBP, SUMO, NusA, Trx | Significant |
Dual-purpose | Both purification and solubility | GST, MBP (with amylose) | Moderate to significant |
A His6 tag does not help your protein fold. Don't expect it to fix solubility problems.
The Big Four: Detailed Comparison
MBP (Maltose Binding Protein)
Size: ~42.5 kDa (370 residues)
Mechanism: MBP is one of the most effective solubility enhancers known. It folds rapidly and independently, creating a "folding nucleus" that promotes passenger protein folding. Its large hydrophilic surface area helps prevent aggregation (Kapust & Waugh, 1999).
The evidence:
Systematic comparison by the Waugh lab showed MBP increased soluble expression for 14 of 18 test proteins (78%), compared to 4 of 18 for GST (22%) and 6 of 18 for Trx (33%)
Structural genomics data confirms MBP as the most broadly effective solubility enhancer
MBP itself is soluble at concentrations exceeding 100 mg/mL
Affinity purification: Amylose resin (binds maltose). Moderate affinity (~1 µM Kd). Works well but resin capacity is lower than Ni-NTA.
Cleavage options: TEV protease site between MBP and passenger is most common. Factor Xa and enterokinase sites also used.
When to use MBP:
First-line choice for "difficult" proteins
When solubility is the primary concern
When you can tolerate the 42 kDa size during early purification
When NOT to use MBP:
When size matters (crystallography of small proteins—MBP may dominate crystal packing)
When you need a native N-terminus (TEV leaves a Ser residue)
When amylose purification is insufficient (consider adding His6)
Common dual-tag strategy: His6-MBP-TEV-target. Purify first on Ni-NTA (high capacity), cleave with TEV, reverse Ni-NTA to remove His6-MBP.
SUMO (Small Ubiquitin-like Modifier)
Size: ~11 kDa (100 residues)
Mechanism: SUMO enhances solubility through a chaperone-like effect and its highly soluble surface. Despite its smaller size, SUMO provides meaningful solubility enhancement—not as strong as MBP, but substantial (Marblestone et al., 2006).
The killer feature: Ulp1/SENP cleavage
SUMO protease (Ulp1 in yeast, SENP in humans) recognizes the tertiary structure of SUMO, not a linear peptide sequence. This means:
Cleavage leaves a completely native N-terminus—no extra residues
Extremely specific—no off-target cleavage
Efficient—typically complete cleavage in 1–2 hours at room temperature
The cleaved SUMO tag can be removed by reverse Ni-NTA (if His6-SUMO was used)
Affinity purification: Typically used as His6-SUMO, relying on the His6 tag for Ni-NTA purification.
When to use SUMO:
When you need a native N-terminus (structural biology, functional studies)
When tag size matters but you still want solubility enhancement
When clean, efficient cleavage is critical
Excellent for proteins destined for crystallization or NMR
When NOT to use SUMO:
In eukaryotic expression systems (host SUMO proteases will cleave prematurely)
When the N-terminus is buried or critical for folding (Ulp1 won't access it)
GST (Glutathione S-Transferase)
Size: ~26 kDa (211 residues)
Mechanism: GST provides moderate solubility enhancement and convenient purification via glutathione-Sepharose. However, its effectiveness as a solubility enhancer is significantly lower than MBP in systematic comparisons (Kapust & Waugh, 1999).
The dimerization issue:
GST naturally forms homodimers. This means your fusion protein is also a dimer. Consequences:
Doubles apparent molecular weight on SEC
May induce artificial dimerization of the passenger protein
Can mask or distort protein-protein interactions
For crystallization: GST dimer may dominate crystal contacts
For some applications, the forced dimerization is actually useful (e.g., studying avidity effects). But for most, it's a complication.
Affinity purification: Glutathione-Sepharose. Good capacity, mild elution (10 mM reduced glutathione). But: reduced glutathione can interfere with downstream assays involving redox chemistry.
Cleavage options: PreScission/3C protease (leaves 5 extra residues) or thrombin (leaves 2 extra residues, but thrombin is less specific).
When to use GST:
When you want easy affinity purification with mild conditions
Pull-down assays (GST pull-down is a standard technique)
When dimerization is acceptable or desired
Quick-and-dirty expression screening
When NOT to use GST:
When you need monomeric protein
When maximum solubility enhancement is critical
For crystallization of the passenger protein
For any assay where dimerization state matters
Thioredoxin (Trx)
Size: ~12 kDa (109 residues)
Mechanism: Thioredoxin promotes cytoplasmic disulfide bond formation when expressed in appropriate E. coli strains (like Origami or SHuffle). It also provides moderate solubility enhancement for small to medium proteins (LaVallie et al., 1993).
The disulfide niche:
Trx is uniquely valuable when your protein requires disulfide bonds:
In standard E. coli cytoplasm, disulfide bonds don't form (reducing environment)
Trx fusion + SHuffle strain enables cytoplasmic disulfide formation
This can replace periplasmic expression (higher yields) or refolding (more reliable)
Affinity purification: No intrinsic affinity tag—typically used as His6-Trx or Trx-Strep.
When to use Trx:
Small proteins (< 25 kDa) with disulfide bonds
When combined with SHuffle or Origami strains
Insulin-like proteins, small cysteine-rich peptides
When tag size must be small
When NOT to use Trx:
Large proteins (Trx's solubility enhancement is limited for large passengers)
Proteins without disulfide bonds (MBP or SUMO would be better choices)
When you need the strongest possible solubility enhancement
Comprehensive Comparison Table
Feature | MBP | SUMO | GST | Thioredoxin | NusA |
|---|---|---|---|---|---|
Size (kDa) | 42.5 | 11 | 26 | 12 | 55 |
Solubility enhancement | Excellent | Good | Moderate | Moderate | Excellent |
Intrinsic purification | Amylose | None (use His6) | Glutathione | None (use His6) | None (use His6) |
Oligomeric state | Monomer | Monomer | Dimer | Monomer | Monomer |
Best cleavage | TEV | Ulp1/SENP | 3C/PreScission | TEV or 3C | TEV |
Native N-terminus | No (Ser from TEV) | Yes (Ulp1) | No (extra residues) | No | No |
Works in eukaryotes | Yes | No (host SENPs) | Yes | Yes | No (bacterial) |
Best for | General solubility | Structural biology | Pull-downs | Disulfide proteins | Last resort difficult |
Cost/construct | Low | Low | Low | Low | Low |
Second-Tier Options Worth Knowing
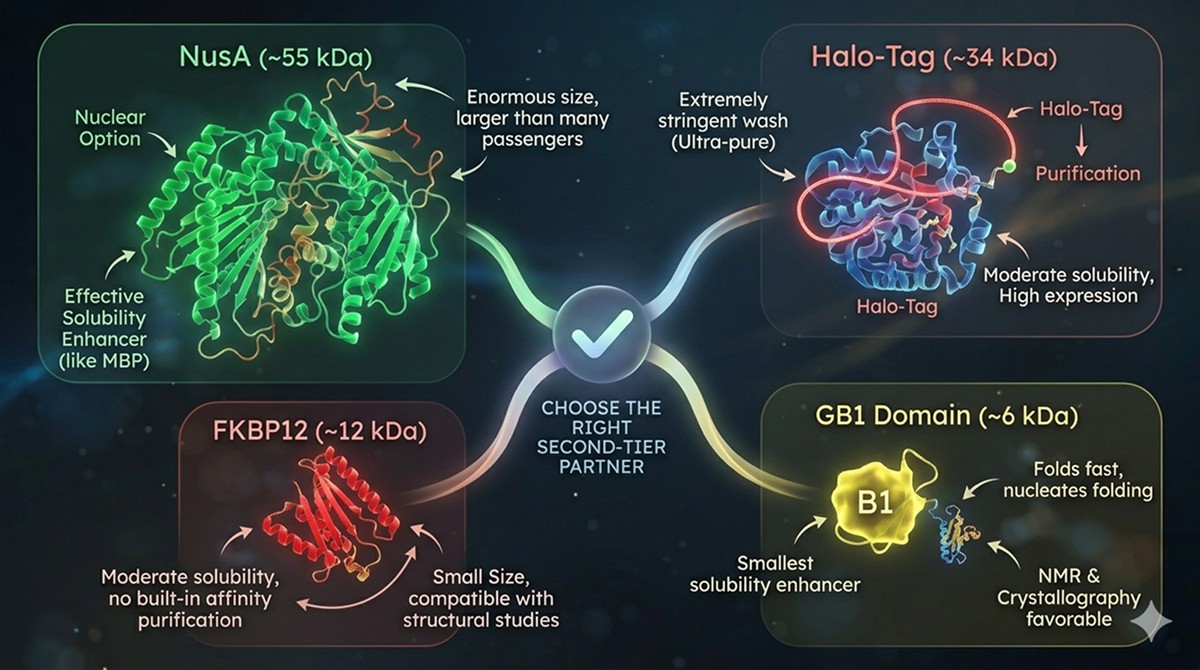
NusA (~55 kDa)
The "nuclear option" for insoluble proteins. NusA is one of the most effective solubility enhancers, rivaling MBP (De Marco et al., 2004). But at 55 kDa, it's enormous—often larger than the passenger protein. Use when MBP fails.
GB1 Domain (~6 kDa)
The smallest effective solubility enhancer. The B1 domain of Streptococcal protein G folds extremely fast and can nucleate folding of small passengers. Limited to small proteins (<30 kDa) but its tiny size makes it attractive for NMR and crystallography (Huth et al., 1997).
FKBP12 (~12 kDa)
Underappreciated option. FKBP fusions provide moderate solubility enhancement and the small size is compatible with structural studies. No built-in affinity purification.
Halo-Tag (~34 kDa)
Engineered from a dehalogenase. Forms a covalent bond with its chloroalkane substrate, enabling extremely stringent wash conditions during purification. High expression levels but moderate solubility enhancement. Best for applications requiring ultra-pure protein.
Decision Tree: Which Fusion Partner for Your Protein?
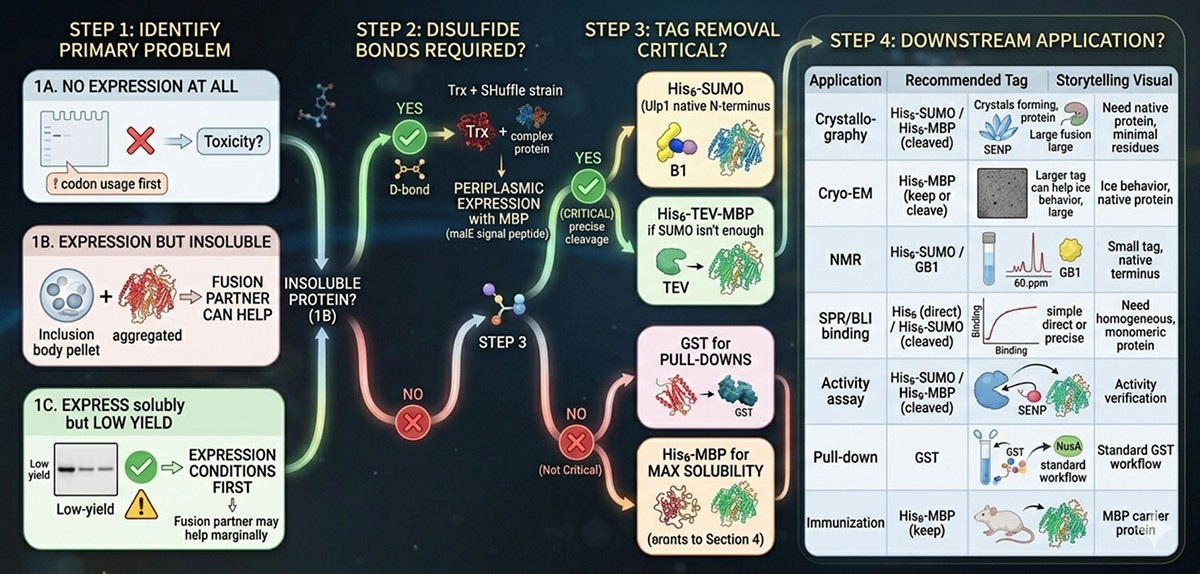
Step 1: What's Your Primary Problem?
Problem: No expression at all
→ Check codon usage first. If codon-optimized and still no expression, the problem may be toxicity. Try low-copy vectors, tight promoters, or autoinduction.
Problem: Expression but insoluble (inclusion bodies)
→ Fusion partner can help. Proceed to Step 2.
Problem: Expresses solubly but low yield
→ Optimize expression conditions first (temperature, inducer concentration, media). Fusion partner may help marginally.
Step 2: Does Your Protein Need Disulfide Bonds?
Yes → Trx + SHuffle strain, or periplasmic expression with MBP (malE signal peptide)
No → Proceed to Step 3
Step 3: How Important Is Tag Removal?
Critical (structural biology, functional assays):
→ His6-SUMO (Ulp1 gives native N-terminus)
→ Or His6-MBP-TEV if SUMO isn't enough for solubility
Not critical (pull-downs, preliminary characterization):
→ GST for pull-down applications
→ His6-MBP for maximum solubility
Step 4: What's Your Downstream Application?
Application | Recommended Tag | Reason |
|---|---|---|
Crystallography | His6-SUMO or His6 + MBP (cleaved) | Need native protein, minimal extra residues |
Cryo-EM | His6-MBP (keep on or cleave) | Large tag can help with ice behavior |
NMR | His6-SUMO or GB1 | Small tag, native terminus |
SPR/BLI binding | His6 (direct) or His6-SUMO (cleaved) | Need homogeneous, monomeric protein |
Activity assay | His6-SUMO or His6-MBP (cleaved) | Verify tag doesn't affect activity |
Pull-down | GST | Standard GST pull-down workflow |
Immunization | His6-MBP (keep on) | MBP can act as carrier protein |
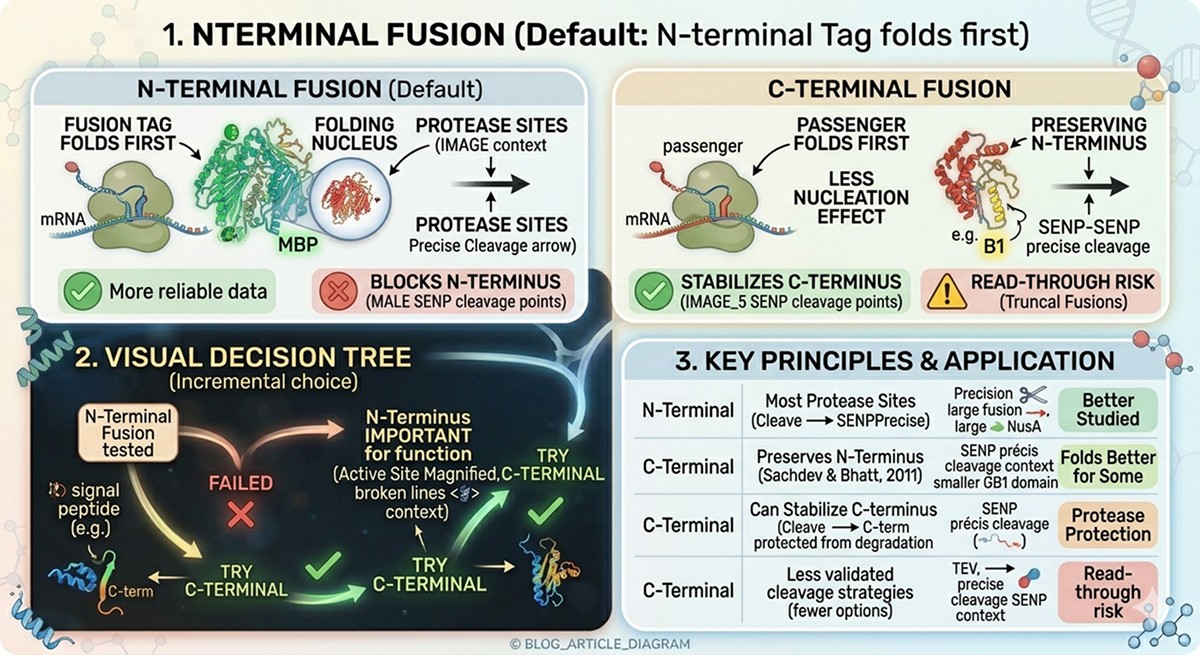
Tag Placement: N-Terminal vs C-Terminal
Most fusion partners are placed at the N-terminus. But C-terminal fusions work better for some proteins.
N-Terminal Fusion (Default)
Advantages:
Fusion partner folds first (translated first), creating a folding nucleus
Most protease cleavage sites are designed for N-terminal fusions
Better studied, more reliable data
Disadvantages:
Blocks the native N-terminus
If the N-terminus is functionally important, the fusion may interfere
C-Terminal Fusion
Advantages:
Preserves native N-terminus
Some proteins fold better with a free N-terminus (Sachdev & Bhatt, 2011)
Can stabilize proteins that are prone to C-terminal degradation
Disadvantages:
Fusion partner folds after the passenger (less folding nucleation effect)
Fewer validated cleavage strategies
Read-through or premature termination can produce truncated fusions
When to Try C-Terminal
N-terminal fusion tested and failed
N-terminus is known to be important for function
Protein has a signal peptide or pro-peptide at the N-terminus
Cleavage Strategies
Protease Comparison
Protease | Recognition Site | Extra Residues After Cleavage | Specificity | Typical Conditions | Cost |
|---|---|---|---|---|---|
TEV | ENLYFQ↓S | 1 (Ser) | Very high | 4°C–RT, 2–16 h | Low (can be expressed in-house) |
3C/PreScission | LEVLFQ↓GP | 2 (Gly-Pro) | High | 4°C, 4–16 h | Moderate |
Ulp1/SENP | SUMO fold (structural) | 0 (native N-term) | Excellent | RT, 0.5–2 h | Low (in-house) |
Thrombin | LVPR↓GS | 2 (Gly-Ser) | Moderate | RT, 1–4 h | Low |
Enterokinase | DDDDK↓ | 0 | Low (can overdigest) | RT, 2–16 h | Moderate |
Factor Xa | IEGR↓ | 0 | Low (can overdigest) | RT, 2–16 h | Moderate |
Recommendation: TEV or Ulp1 for most applications. They are the most specific and reliable. Avoid thrombin, enterokinase, and Factor Xa unless your lab has established protocols—their lower specificity frequently causes problems.
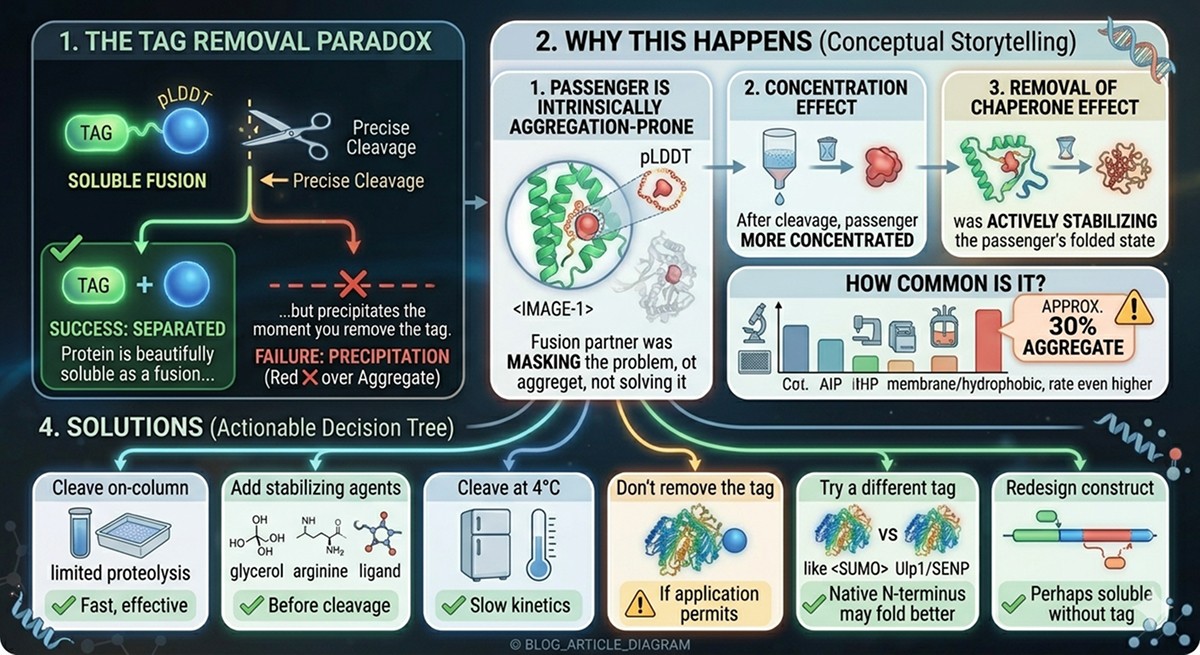
The Tag Removal Paradox
One of the most frustrating problems in fusion protein work: your protein is beautifully soluble as a fusion but precipitates the moment you remove the tag.
Why This Happens
The passenger protein is intrinsically aggregation-prone: The fusion partner was masking the problem, not solving it
Concentration effect: After cleavage and reverse purification, the passenger is more concentrated
Removal of chaperone effect: The fusion partner was actively stabilizing the passenger's folded state
How Common Is It?
Estimates suggest that approximately 30% of proteins that express solubly as fusions precipitate after tag removal. For membrane-associated or highly hydrophobic proteins, the rate is even higher.
Solutions
Cleave on-column at low concentration
Add stabilizing agents before cleavage (glycerol, arginine, specific ligands)
Cleave at 4°C to slow aggregation kinetics
Don't remove the tag if your application permits it
Try a different tag: sometimes SUMO fusion proteins survive cleavage better than MBP fusions (the native N-terminus may fold better)
Redesign the construct: perhaps a shorter construct (removing disordered regions) will be soluble without the tag
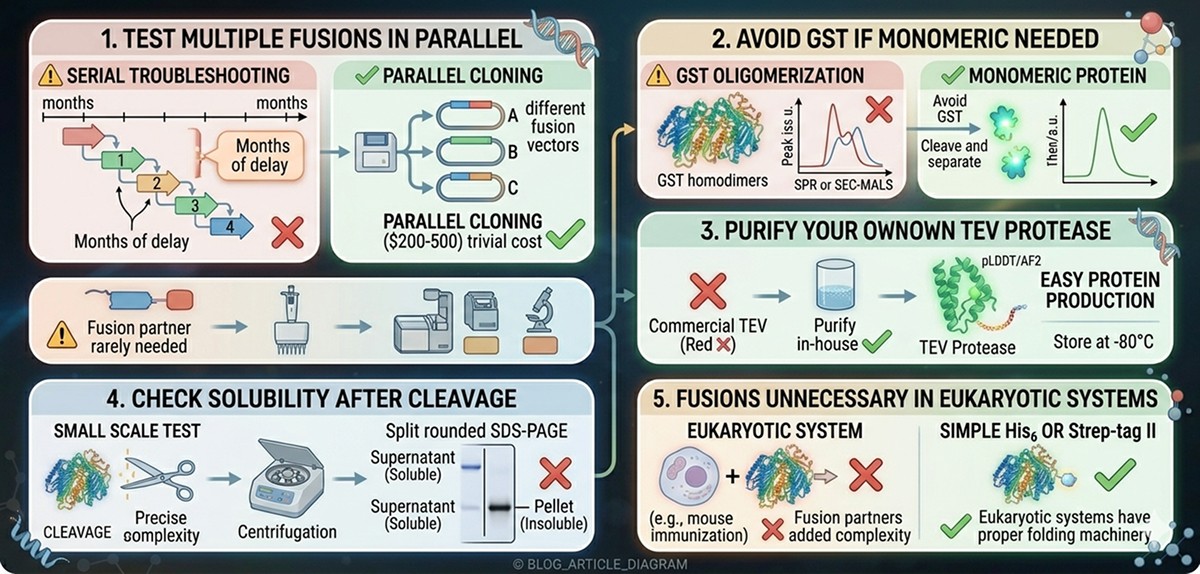
Common Mistakes and Troubleshooting
Mistake 1: Not Testing Multiple Fusions in Parallel
The fix: Clone your gene into 2–3 different fusion vectors simultaneously. The cost of parallel cloning (~$200–500) is trivial compared to months of serial troubleshooting.
Mistake 2: Using GST When You Need Monomeric Protein
The fix: If your downstream assay is sensitive to oligomeric state (SEC-MALS, AUC, SPR), avoid GST or plan to cleave and separate.
Mistake 3: Forgetting That TEV Protease Itself Needs Purification
The fix: Express and purify your own TEV protease. It's one of the easiest proteins to produce in E. coli. Store aliquots at –80°C. Never buy commercial TEV if you have basic protein purification capabilities.
Mistake 4: Not Checking for Solubility AFTER Cleavage
The fix: Always run a small-scale cleavage test before scaling up. Check solubility of the cleaved product by centrifugation + SDS-PAGE of supernatant vs pellet.
Mistake 5: Using Fusion Partners in Eukaryotic Systems That Don't Need Them
The fix: Insect and mammalian cells have proper folding machinery. Fusion partners are rarely needed and add unnecessary complexity. Use a simple His6 or Strep-tag.
The Bottom Line
Situation | First Choice | Second Choice | Avoid |
|---|---|---|---|
General insolubility | His6-MBP | His6-SUMO | His6 alone (no solubility help) |
Structural biology (need native N-term) | His6-SUMO | His6-MBP + TEV | GST (dimerization) |
Disulfide bonds needed | His6-Trx + SHuffle | Periplasmic MBP | Cytoplasmic expression without Trx |
Pull-down assays | GST | His6-MBP | — |
"Nothing else works" | His6-NusA | MBP-SUMO tandem | Giving up before trying NusA |
Small protein (<15 kDa) | His6-SUMO or GB1 | His6-Trx | Large tags (NusA, MBP) may overwhelm |
Eukaryotic expression | His6 or Strep-tag | — | SUMO (host SENPs will cleave) |
The fusion partner is not a magic bullet—it won't rescue a protein that needs PTMs, cofactors, or a specific membrane environment. But for proteins that simply struggle to fold in E. coli, the right fusion partner can be the difference between an empty gel lane and milligrams of pure protein.
Rational Fusion Partner Selection with Orbion
Orbion's Construct Design module includes a component library of purification tags and fusion partners, allowing you to assemble expression-ready constructs with defined boundaries, tags, and codon optimization in one workflow. AstraSUIT predicts expression system suitability—including host association, cofactor requirements, and membrane type—helping you determine whether E. coli + fusion partner is even the right strategy, or whether you should go directly to insect or mammalian cells.
AstraPTM predictions flag PTM requirements (glycosylation, disulfide bonds) that directly affect tag choice: if your protein needs N-glycosylation, no E. coli fusion partner will help. Combined with the Bench module for protocol generation, you can go from "my protein doesn't express" to a rationally designed construct with matched protocols in a single workflow.
References
Kapust RB & Waugh DS. (1999). Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Science, 8(8):1668-1674. PMC22049
Esposito D & Chatterjee DK. (2006). Enhancement of soluble protein expression through the use of fusion tags. Current Opinion in Biotechnology, 17(4):353-358. PMC2706091
Marblestone JG, et al. (2006). Comparison of SUMO fusion technology with traditional gene fusion systems: enhanced expression and solubility with SUMO. Protein Science, 15(1):182-189. PMC3489955
LaVallie ER, et al. (1993). A thioredoxin gene fusion expression system that circumvents inclusion body formation in the E. coli cytoplasm. Nature Biotechnology, 11:187-193. Link
De Marco V, et al. (2004). Bacteria co-transformed with recombinant proteins and chaperones cloned in independent plasmids are suitable for expression tuning. Journal of Biotechnology, 109(1-2):45-52. PMC515306
Huth JR, et al. (1997). Design of an expression system for detecting folded protein domains and mapping macromolecular interactions by NMR. Protein Science, 6(11):2359-2364. PMC2143601
Malhotra A. (2009). Tagging for protein expression. Methods in Enzymology, 463:239-258. Link
Waugh DS. (2005). Making the most of affinity tags. Trends in Biotechnology, 23(6):316-320. Link
Costa S, et al. (2014). Fusion tags for protein solubility, purification and immunogenicity in Escherichia coli: the novel Fh8 system. Frontiers in Microbiology, 5:63. PMC3933005
Butt TR, et al. (2005). SUMO fusion technology for difficult-to-express proteins. Protein Expression and Purification, 43(1):1-9. Link
Book a 20-Minute Demo
Sign up free for unlimited Overview runs — summary, sequence-based analysis, homology search. For the full Characterization — PTMs, binding sites, stability variants, construct design — book a demo and we'll run your target live.
