Blog
Orbion Team
What pLDDT and PAE Actually Tell You (And What They Don't)

Your AlphaFold model just finished. The average pLDDT is 85—looks great. You download the structure, open it in PyMOL, and start planning mutations at the active site. But zoom in on that loop near residue 140. pLDDT drops to 35. That 200-residue C-terminal domain? PAE says it's floating in space relative to the catalytic core. Your "85 average" is hiding the fact that half your structure is unreliable—and you're about to design experiments based on the wrong parts.
AlphaFold's confidence scores are the most valuable—and most misunderstood—outputs of the model. Knowing how to read them separates researchers who waste months on artifact-based hypotheses from those who design the right experiment the first time.
Key Takeaways
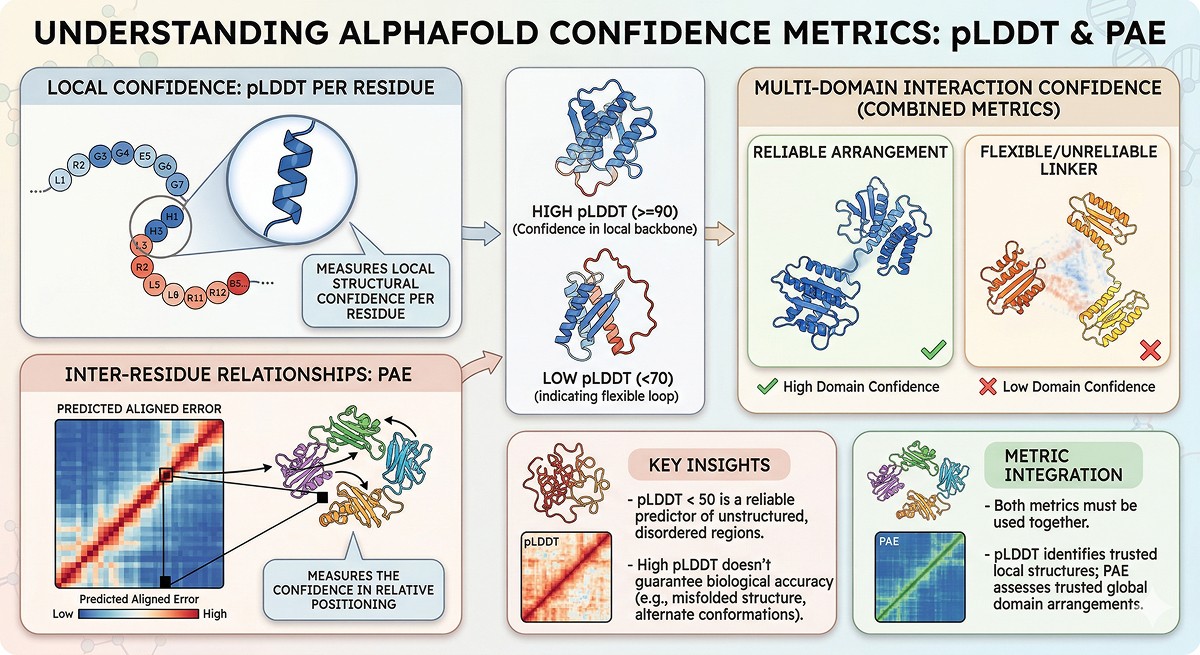
pLDDT measures local structural confidence per residue, not global model accuracy or biological relevance
PAE measures the confidence in relative positioning between any two residues—essential for multi-domain proteins
pLDDT < 50 is a reliable disorder predictor, often outperforming dedicated disorder prediction tools
High pLDDT doesn't guarantee biological accuracy: a confidently predicted structure can still be wrong if the biology differs from the training data
Use both metrics together: pLDDT tells you which residues to trust locally; PAE tells you which domain arrangements to trust globally
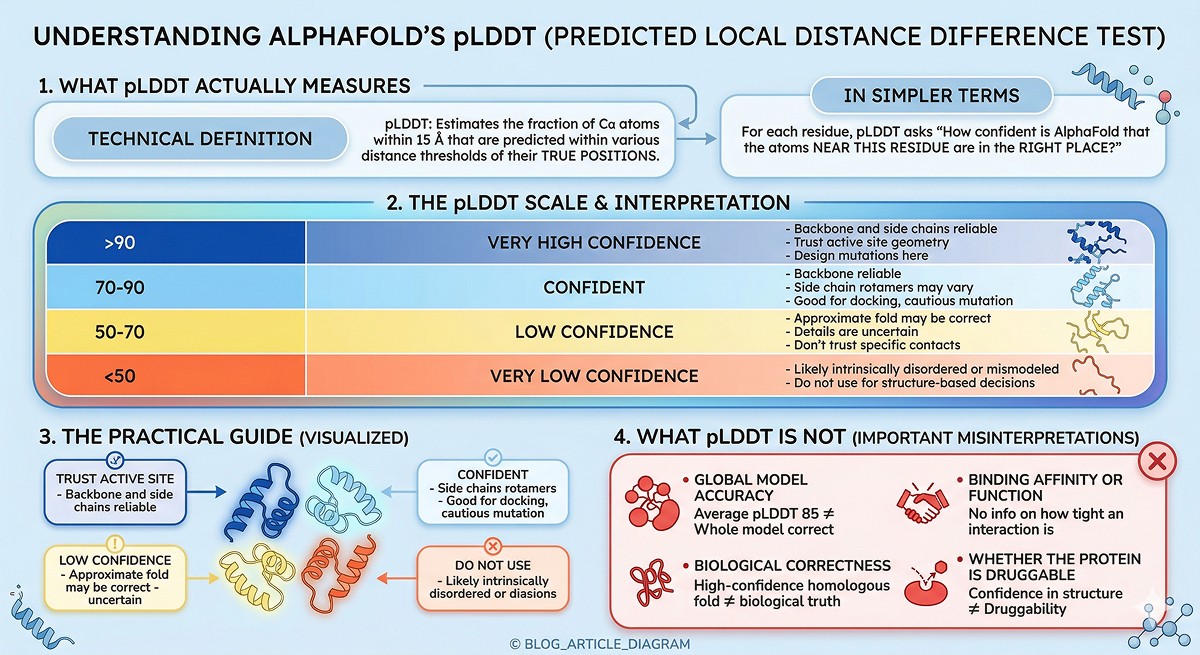
What pLDDT Actually Measures
The Technical Definition
pLDDT stands for predicted Local Distance Difference Test. It estimates how well AlphaFold predicts the local atomic environment around each residue—specifically, the fraction of Cα atoms within 15 Å that are predicted within various distance thresholds of their true positions (Jumper et al., 2021).
In simpler terms: for each residue, pLDDT asks "how confident is AlphaFold that the atoms near this residue are in the right place?"
The Scale
pLDDT Range | Color (Default) | Interpretation | Practical Meaning |
|---|---|---|---|
> 90 | Deep blue | Very high confidence | Backbone and side chains reliable. Trust active site geometry, design mutations here. |
70–90 | Light blue | Confident | Backbone reliable. Side chain rotamers may vary. Good for docking, cautious mutation design. |
50–70 | Yellow | Low confidence | Approximate fold may be correct, but details are uncertain. Don't trust specific contacts. |
< 50 | Orange/red | Very low confidence | Likely intrinsically disordered or mismodeled. Do not use for structure-based decisions. |
What pLDDT Is NOT
pLDDT is frequently misinterpreted. It does not measure:
Global model accuracy: A model with average pLDDT of 85 can have entire domains that are wrong
Biological correctness: A high-confidence prediction of a homologous fold doesn't mean your protein actually adopts that fold
Binding affinity or function: pLDDT says nothing about whether a binding site works or how tight an interaction is
Whether the protein is druggable: Confidence in structure ≠ confidence in druggability
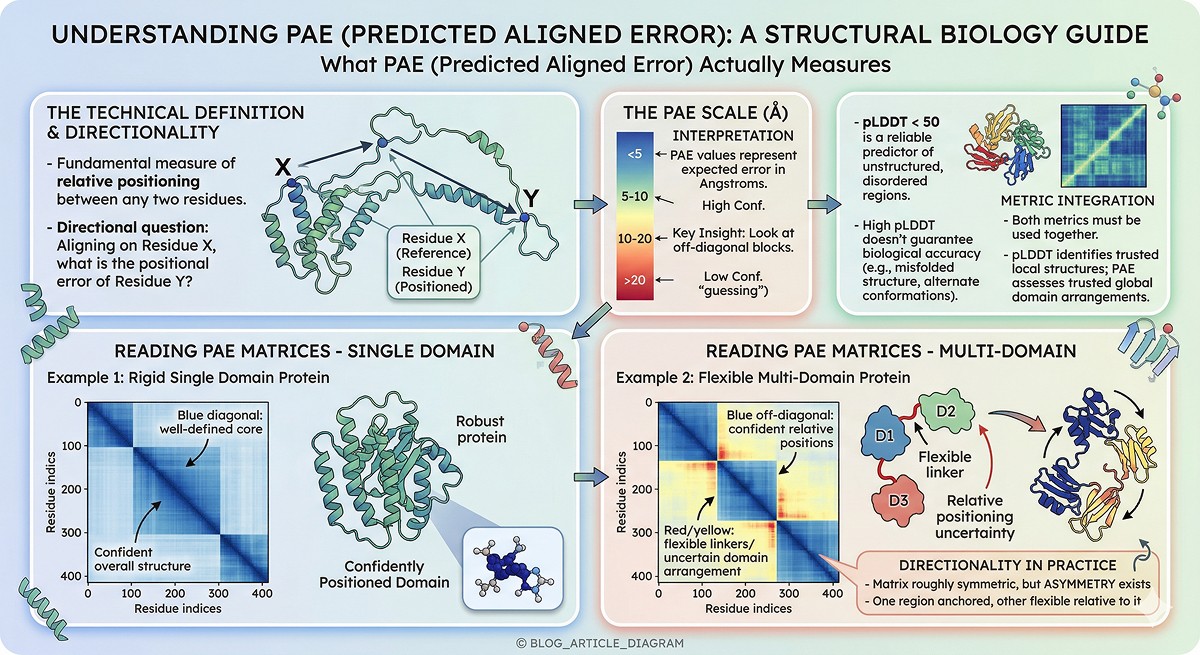
What PAE Actually Measures
The Technical Definition
PAE (Predicted Aligned Error) answers a different question: if you align the structure using residue X as the reference, what is the expected positional error of residue Y? (Evans et al., 2022)
This is fundamentally about relative positioning—not local confidence.
The Scale
PAE (Å) | Interpretation |
|---|---|
< 5 | Very high confidence in relative position |
5–10 | High confidence |
10–20 | Moderate confidence—positions are approximate |
> 20 | Low confidence—AlphaFold is essentially guessing the relative arrangement |
How to Read PAE Matrices
The PAE matrix is a 2D heatmap (residue × residue):
Blue blocks along the diagonal: Rigid, well-defined structural domains
Blue off-diagonal blocks: Two regions whose relative positioning is confident (rigid body relationship)
Red/yellow between blocks: Flexible linkers, uncertain domain arrangements, or genuine conformational heterogeneity
Key insight: PAE is directional. Aligning on residue 50 and asking about residue 200 can give a different answer than aligning on 200 and asking about 50. In practice, for well-folded proteins the matrix is roughly symmetric, but asymmetry indicates that one region is "anchored" while the other is flexible relative to it.
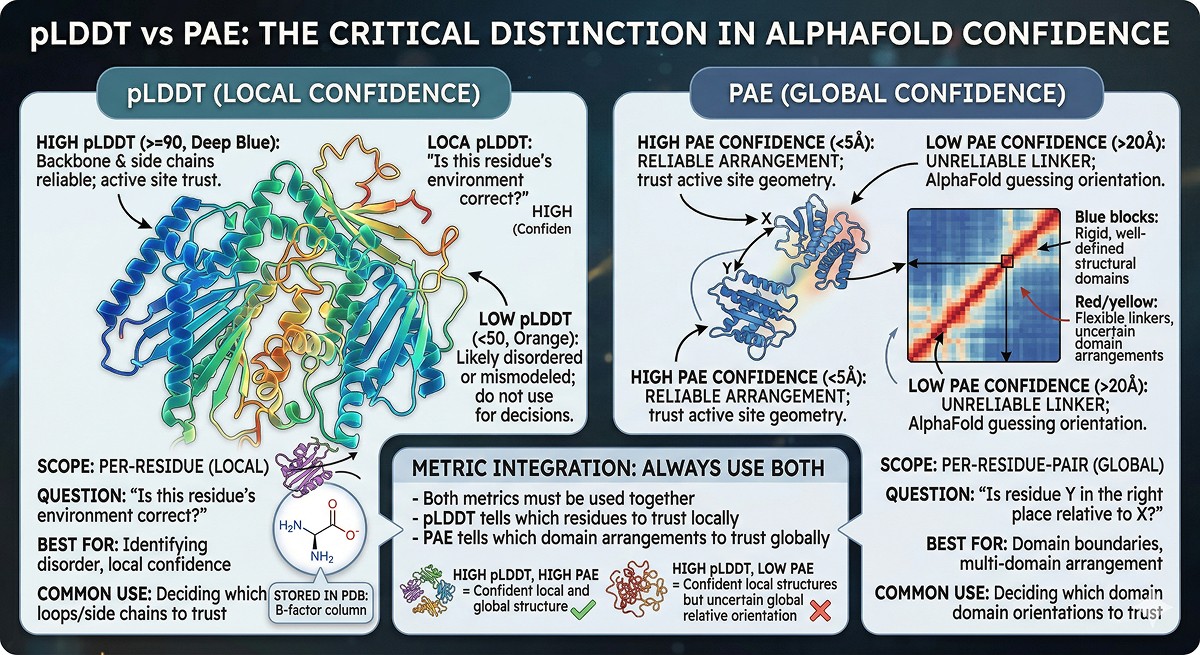
PAE vs pLDDT: The Critical Distinction
Feature | pLDDT | PAE |
|---|---|---|
Scope | Per-residue (local) | Per-residue-pair (global) |
Question | "Is this residue's environment correct?" | "Is residue Y in the right place relative to X?" |
Best for | Identifying disorder, local confidence | Domain boundaries, multi-domain arrangement |
Common use | Deciding which loops/side chains to trust | Deciding which domain orientations to trust |
Stored in PDB | B-factor column | Separate JSON file |
pLDDT as a Disorder Predictor
One of the most practically useful (and underappreciated) features of pLDDT is its strong correlation with intrinsic disorder.
The Evidence
Ruff and Pappu (2021) showed that AlphaFold's pLDDT scores correlate strongly with intrinsic disorder. Regions with pLDDT < 50 correspond closely to experimentally validated intrinsically disordered regions (IDRs).
Wilson et al. (2022) systematically compared AlphaFold pLDDT to dedicated disorder predictors (IUPred2, PONDR-VSL2, ESpritz) and found that pLDDT < 50 performs comparably or better for identifying long disordered regions (>30 residues).
Why This Matters
If your AlphaFold model has a 100-residue stretch with pLDDT < 50, that region is almost certainly disordered. This has immediate practical consequences:
Don't trust the structure in that region—AlphaFold is essentially placing a random extended chain
Consider removing it for crystallization or cryo-EM (construct design)
Expect problems with aggregation, proteolysis, and heterogeneity if you include it
It may be functional: disordered regions mediate many protein-protein interactions and are often regulatory
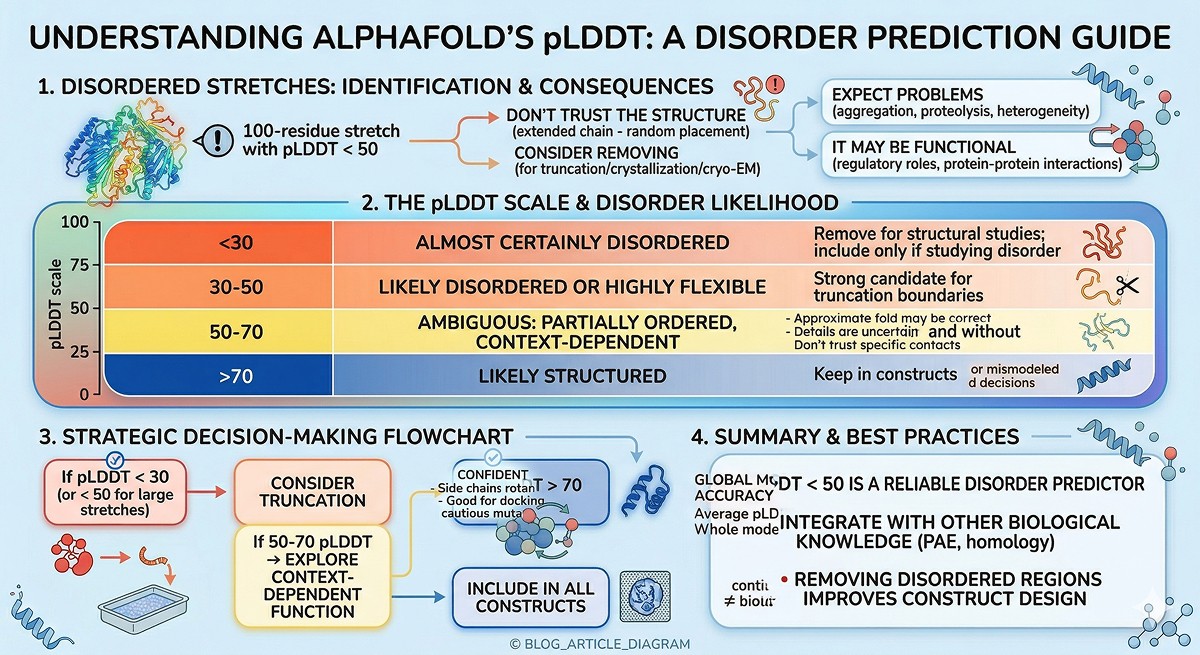
pLDDT Thresholds for Disorder
pLDDT | Disorder Likelihood | Action |
|---|---|---|
< 30 | Almost certainly disordered | Remove for structural studies; include only if studying disorder |
30–50 | Likely disordered or highly flexible | Strong candidate for truncation boundaries |
50–70 | Ambiguous: may be partially ordered, context-dependent | Test both with and without |
> 70 | Likely structured | Keep in constructs |
PAE for Domain Boundary Detection
Reading Domain Architecture from PAE
The PAE matrix is arguably the best tool available for identifying domain boundaries in predicted structures—even better than visual inspection of the 3D model.
What to look for:
Blue square blocks along the diagonal: Each block is a rigid structural domain
Sharp transitions from blue to red/yellow: Domain boundaries where flexible linkers connect rigid units
Off-diagonal blue patches: Domains that are rigidly positioned relative to each other (e.g., in a fixed multi-domain arrangement)
Large red/yellow off-diagonal regions: Domains whose relative orientation is uncertain—AlphaFold predicts each domain's fold but not how they sit together
From PAE to Construct Design
PAE Pattern | What It Means | Construct Design Implication |
|---|---|---|
Single blue block (full length) | Single rigid domain | Express full-length |
Two blue blocks, blue between | Two-domain protein, fixed arrangement | Express full-length or individual domains |
Two blue blocks, red between | Two domains, flexible linker | Express domains separately for crystallization |
Blue block + red C-terminus | Structured domain + disordered tail | Truncate the disordered tail |
Fragmented small blue patches | Multi-domain or mostly disordered | Careful boundary design needed; limited proteolysis may help |
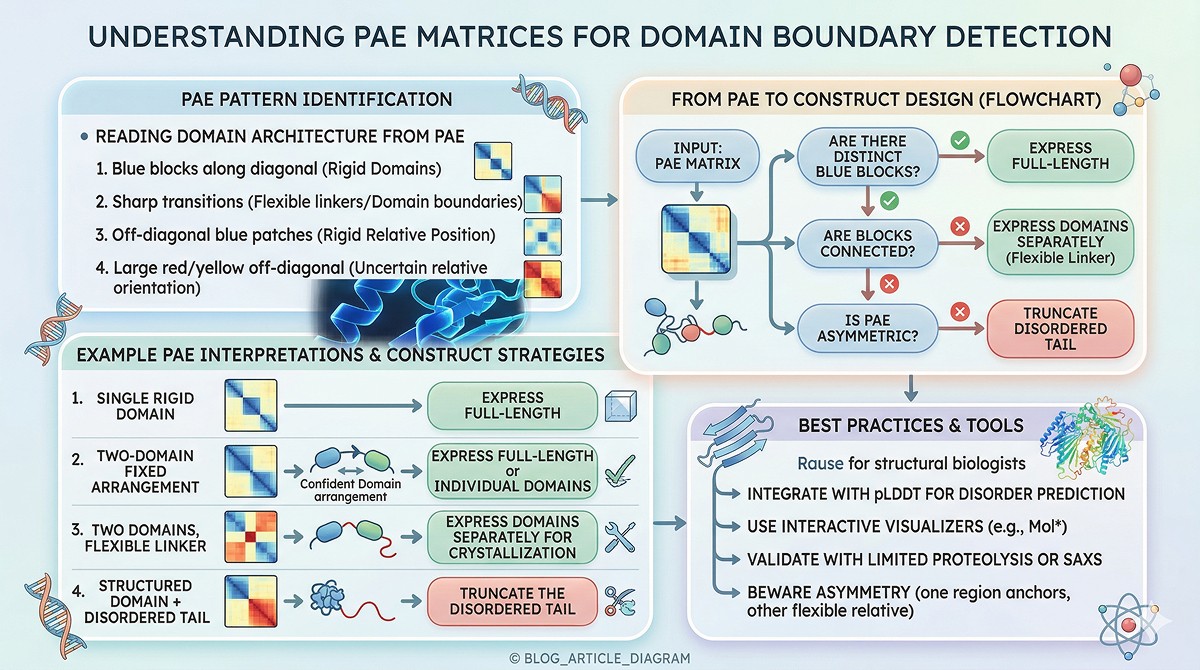
Practical Example
A 450-residue protein with this PAE pattern:
Residues 1–30: Red (low pLDDT, disordered N-terminus)
Residues 31–200: Blue block (Domain 1)
Residues 201–220: Red stripe (flexible linker)
Residues 221–400: Blue block (Domain 2)
Residues 401–450: Red (disordered C-terminus)
Construct strategy:
Full-length for functional studies
Residues 25–205 for Domain 1 crystallization
Residues 215–405 for Domain 2 crystallization
Residues 25–405 for the two-domain construct (may not crystallize if linker is truly flexible)
When High pLDDT Is Misleading
High confidence doesn't always mean high accuracy. Here are the traps.
Trap 1: Homology-Driven Confidence on a Wrong Fold
AlphaFold learns from known structures. If your protein is homologous to well-characterized proteins, AlphaFold will confidently predict a similar fold—even if your protein has diverged functionally or structurally.
Example: A bacterial toxin homologous to a well-studied enzyme family. AlphaFold predicts the enzyme fold with pLDDT > 85. But the toxin has evolved a completely different active site geometry. The fold is "right" (similar to homologs) but the functional details are wrong.
Trap 2: Missing Cofactors and Ligands
AlphaFold predicts the apo structure. If your protein requires a metal ion, cofactor, or ligand for its native conformation, the predicted structure may be confidently wrong in the binding region.
Example: A metalloprotease with a zinc-binding site. AlphaFold predicts the overall fold with high pLDDT but the zinc-coordinating residues may not be in the correct geometry because there's no zinc in the prediction.
Trap 3: Chimeric or Engineered Constructs
If you submit a chimeric protein (e.g., a fusion construct, circular permutant, or engineered variant), AlphaFold will try to fold each part based on its training data. It may confidently predict individual domains but place them incorrectly relative to each other.
Trap 4: Conformational States
AlphaFold typically predicts one state. Many proteins exist in multiple conformations (open/closed, active/inactive). The predicted state may have high pLDDT but represent only one of several biologically relevant forms.
Notable example: GPCRs—AlphaFold often predicts the inactive state with high confidence, but the active state (bound to agonist and G-protein) looks very different (Heo & Bhatt, 2022).
When Low pLDDT Doesn't Mean "Wrong"
The inverse trap: assuming low pLDDT means a bad prediction.
Case 1: Genuine Disorder
Low pLDDT often means the region IS intrinsically disordered. AlphaFold isn't wrong—it's correctly telling you there's no stable structure to predict. This is a feature, not a bug.
Case 2: Conformational Flexibility
Loops that sample multiple conformations will have low pLDDT because no single conformation dominates. The "average" position AlphaFold predicts may be close to any individual state without matching any perfectly.
Case 3: Crystal Packing Artifacts
Some regions that are well-ordered in crystal structures are disordered in solution. AlphaFold predicts solution behavior, so it may assign low pLDDT to regions stabilized only by crystal contacts. Neither is "wrong"—they're answering different questions.
Case 4: Orphan Proteins
For proteins with few homologs, AlphaFold has less evolutionary information. Lower pLDDT may reflect insufficient data rather than genuine disorder. These proteins may actually be well-structured but poorly predicted.
The Practical Decision Table
This is what most researchers actually need: "Given my confidence scores, can I do X?"
Research Decision | Minimum pLDDT | PAE Requirement | Notes |
|---|---|---|---|
Trust active site geometry for docking | > 85 at active site residues | < 5 Å within active site | Verify with experimental structure if available |
Design point mutations for stability | > 70 at mutation site | < 10 Å for the local domain | Consider if region is buried vs exposed |
Trust a binding site prediction | > 80 at predicted site | < 8 Å within binding pocket | Cross-validate with conservation |
Use structure for molecular dynamics | > 70 overall for the domain | Domain should be a single blue PAE block | Low-pLDDT tails/loops will need special treatment |
Define construct boundaries | Identify where pLDDT drops below 50 | Use PAE to find domain blocks | Cut at disorder-order transitions |
Trust domain arrangement | pLDDT is insufficient | Inter-domain PAE < 10 Å | If PAE is high, domains may be correct individually but wrong relative to each other |
Dock a small molecule | > 80 in binding pocket | < 5 Å within pocket | Side chain positions may still be imperfect |
Design interface mutations (complex) | > 80 at interface | Inter-chain PAE < 10 Å | See separate complex confidence guide |
Trust loop conformation | > 70 for the loop | < 10 Å to anchoring residues | Loops are often the least reliable part |
Homology model for distant homolog | > 60 overall | Single domain PAE block | Lower bar because any model is better than none |
Ten Common Mistakes Researchers Make
1. Averaging pLDDT Across the Whole Protein
A 500-residue protein with 300 residues at pLDDT 95 and 200 at pLDDT 40 has an "average" of 73. That average hides both the excellent core and the terrible tail. Always look at the per-residue profile, not the average.
2. Ignoring PAE Entirely
Most researchers check pLDDT and stop. For any multi-domain protein, PAE is the more important metric for understanding overall architecture.
3. Trusting Loop Conformations at pLDDT 60–70
This "yellow zone" is where AlphaFold is uncertain. Loop conformations in this range may look reasonable in the 3D viewer but could easily be several angstroms off. Don't design experiments that depend on specific contacts in these regions.
4. Treating pLDDT as a Probability
pLDDT 80 does not mean "80% chance of being correct." It's a predicted lDDT score—a measure of local distance accuracy. The relationship between pLDDT and actual accuracy is nonlinear and context-dependent.
5. Assuming One Model = One Truth
AlphaFold generates multiple models (typically 5) and ranks them. The top model isn't always best everywhere. Different models may be better for different regions. Check all ranked models when making critical decisions.
6. Designing Mutations at Domain Interfaces Without Checking PAE
If two domains have high inter-domain PAE (> 15 Å), their relative orientation is uncertain. Any mutation designed at their interface is based on an unreliable arrangement.
7. Forgetting That AlphaFold Predicts Apo Structures
No ligands, no cofactors, no post-translational modifications, no binding partners. The structure you see is the protein alone. Binding sites may rearrange upon ligand binding.
8. Using Low pLDDT to Dismiss Functional Regions
Some of the most biologically important regions—activation loops, regulatory segments, binding motifs—are flexible and show low pLDDT. Low confidence ≠ low importance.
9. Not Considering MSA Depth
AlphaFold's confidence depends heavily on the depth and diversity of the multiple sequence alignment. A protein from an underrepresented organism may have lower pLDDT not because it's disordered, but because AlphaFold lacks evolutionary data. Check MSA depth when interpreting unusual confidence patterns.
10. Comparing pLDDT Across Different Proteins
pLDDT 80 for a well-studied kinase and pLDDT 80 for an orphan protein from an extremophile don't carry the same information. Context matters—MSA depth, fold novelty, and available homologous structures all affect what a given pLDDT score means.
Using Confidence to Choose Experimental Methods
When your AlphaFold model has regions of varying confidence, use this to decide which experiments to pursue.
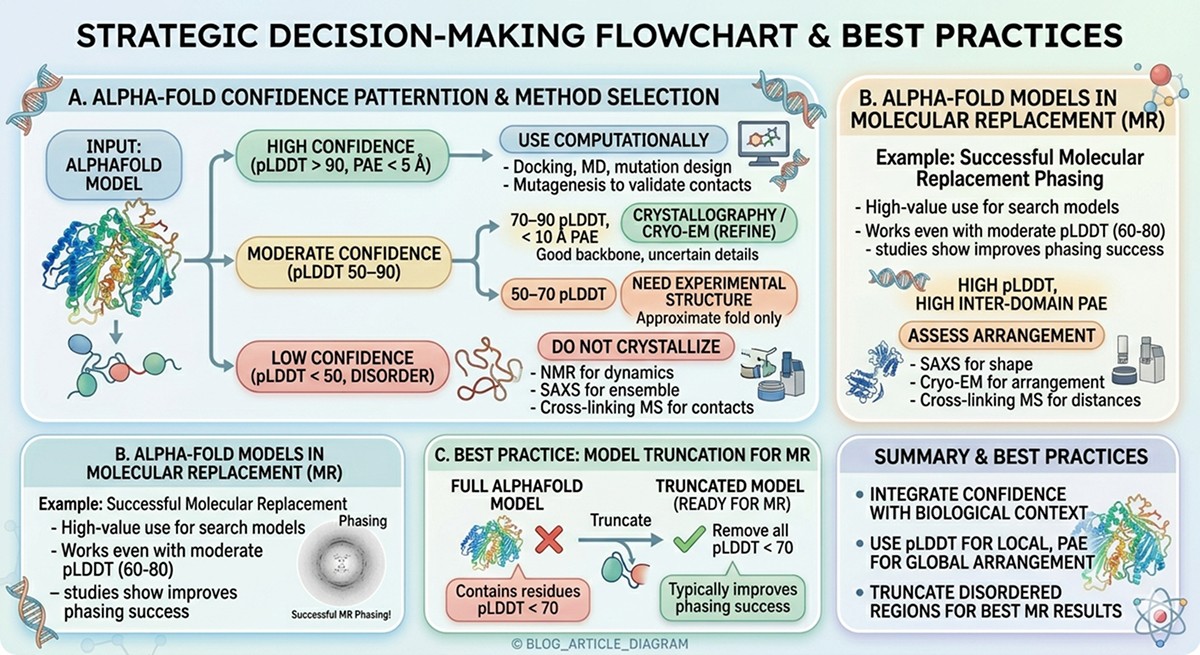
Method Selection by Confidence
Region Confidence | What You Know | Best Experimental Approach |
|---|---|---|
pLDDT > 90, PAE < 5 Å | High-confidence structure | Use computationally: docking, MD, mutation design. Validate key contacts with mutagenesis. |
pLDDT 70–90, PAE < 10 Å | Good backbone, uncertain details | Crystallography or cryo-EM to refine. Use AlphaFold as molecular replacement model. |
pLDDT 50–70 | Approximate fold only | Need experimental structure. AlphaFold model useful for phasing in crystallography. |
pLDDT < 50 (disorder) | No stable structure | NMR for dynamics, SAXS for ensemble, cross-linking MS for contacts. Don't try to crystallize. |
High pLDDT, high inter-domain PAE | Domains correct, arrangement unknown | SAXS for shape, cryo-EM for arrangement, cross-linking MS for distance restraints. |
AlphaFold as Molecular Replacement Model
One of the highest-value uses of AlphaFold models is as search models for X-ray crystallographic molecular replacement. Studies show that AlphaFold models succeed as MR search models even when pLDDT is moderate (60-80), and truncating low-pLDDT regions improves success rates.
Best practice: Remove all residues with pLDDT < 70 before using as an MR model. This typically improves phasing success (McCoy et al., 2022).
Advanced: Combining pLDDT and PAE
The most informative interpretation comes from reading both metrics together.
pLDDT | PAE | Interpretation |
|---|---|---|
High (>80) | Low (<10 Å) | Best case. Structure is reliable locally and globally. |
High (>80) | High (>20 Å) | Individual domains are correct, but relative arrangement is wrong. Common in multi-domain proteins with flexible linkers. |
Low (<50) | Low (<10 Å) | Rare. May indicate a region that's flexible but constrained in position (e.g., a disordered loop between two rigid anchors). |
Low (<50) | High (>20 Å) | Disordered and positionally uncertain. AlphaFold is confident this region has no stable structure AND no fixed position. |
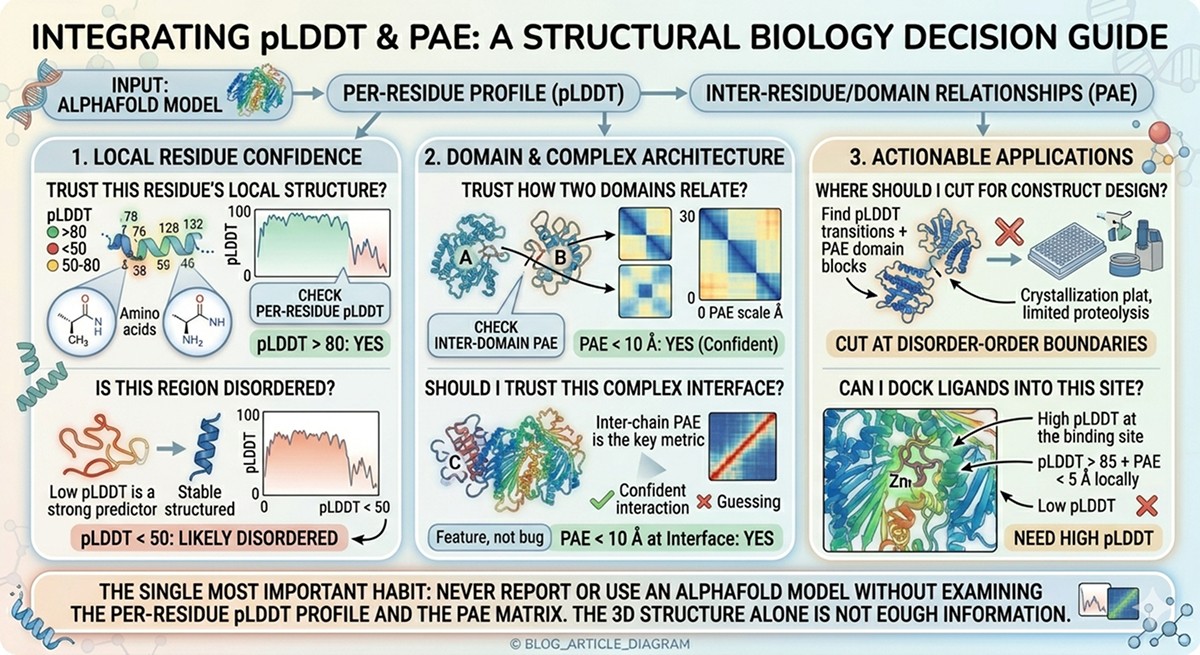
The Bottom Line
Question | Answer | Which Metric |
|---|---|---|
"Can I trust this residue's local structure?" | Check per-residue pLDDT | pLDDT > 80: yes |
"Can I trust how two domains relate?" | Check inter-domain PAE | PAE < 10 Å: yes |
"Is this region disordered?" | Low pLDDT is a strong predictor | pLDDT < 50: likely disordered |
"Where should I cut for construct design?" | Find pLDDT transitions + PAE domain blocks | Cut at disorder-order boundaries |
"Can I dock ligands into this site?" | Need high pLDDT at the binding site | pLDDT > 85 + PAE < 5 Å locally |
"Should I trust this complex interface?" | Inter-chain PAE is the key metric | PAE < 10 Å at interface |
The single most important habit: never report or use an AlphaFold model without examining the per-residue pLDDT profile and the PAE matrix. The 3D structure alone is not enough information.
Interpreting Confidence in Practice
For researchers working with AlphaFold outputs, platforms like Orbion streamline confidence interpretation. The 3D structure viewer supports pLDDT coloring directly, and annotated PDB exports include pLDDT in the B-factor column for use in molecular replacement or downstream tools. The PAE Insight Engine goes further—automatically deriving domain boundaries, hinge regions, and conformational heterogeneity cues from the PAE matrix, so you don't have to interpret raw heatmaps manually.
This matters especially at the construct design stage: when you can see exactly where disorder starts, where domains end, and which linkers are flexible, you can define expression construct boundaries that avoid the most common failure modes. Combining pLDDT visualization with AstraUNFOLD disorder predictions and AstraBIND binding site analysis gives a complete picture—which parts of your protein to trust structurally, which to truncate, and which to investigate experimentally.
References
Jumper J, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596:583-589. Link
Evans R, et al. (2022). Protein complex prediction with AlphaFold-Multimer. bioRxiv. Link
Ruff KM & Pappu RV. (2021). AlphaFold and Implications for Intrinsically Disordered Proteins. Journal of Molecular Biology, 433(20):167208. Link
Wilson CJ, et al. (2022). AlphaFold2 and disorder: Are predicted confidence scores a good substitute for disorder predictors? Proteins, 90(12):2109-2118. Link
Mariani V, et al. (2013). lDDT: a local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics, 29(21):2722-2728. PMC3799472
McCoy AJ, et al. (2022). Implications of AlphaFold2 for crystallographic phasing by molecular replacement. Acta Crystallographica Section D, 78(1):1-13. PMC8662965
Heo L & Bhatt S. (2022). Assessment of AlphaFold predictions of GPCR structures. bioRxiv. PMC9348835
Tunyasuvunakool K, et al. (2021). Highly accurate protein structure prediction for the human proteome. Nature, 596:590-596. Link
Varadi M, et al. (2022). AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Research, 50(D1):D439-D444. PMC8728224
Akdel M, et al. (2022). A structural biology community assessment of AlphaFold2 applications. Nature Structural & Molecular Biology, 29:1056-1067. Link
Book a 20-Minute Demo
Sign up free for unlimited Overview runs — summary, sequence-based analysis, homology search. For the full Characterization — PTMs, binding sites, stability variants, construct design — book a demo and we'll run your target live.
