Blog
Orbion Team
Why Your Protein Loses Activity After Purification (Even Though It's Pure)

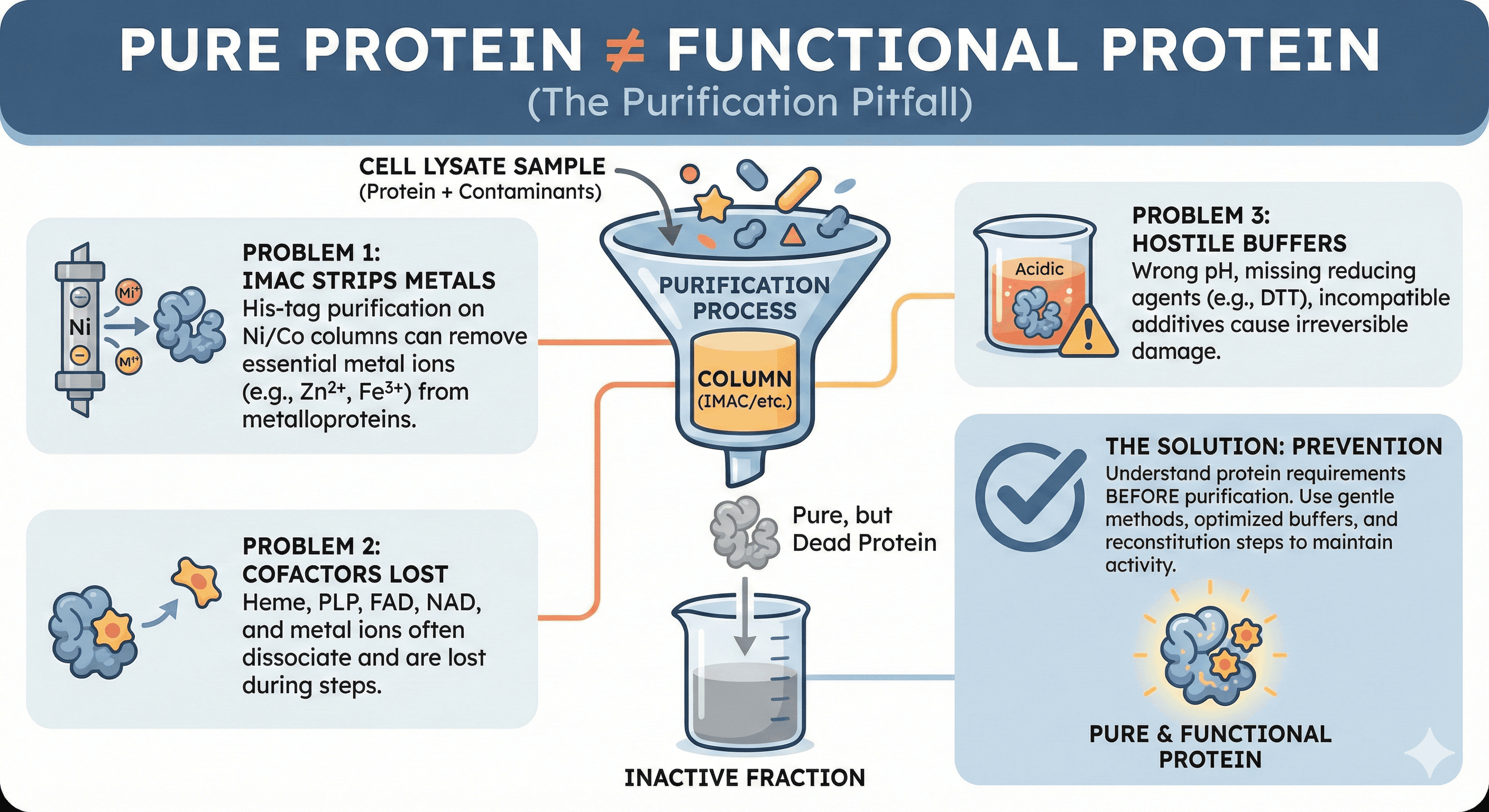
The chromatogram looked perfect. You ran the gel—single band at the expected molecular weight. The mass spec confirmed the identity. Your protein is >95% pure. Then you ran the activity assay. Nothing. The enzyme that was robustly active in crude lysate is now completely dead.
You didn't lose purity. You lost function. And the purification itself was the culprit.
Key Takeaways
Pure protein is not the same as functional protein: Purification removes more than just contaminants—it can strip essential cofactors, metal ions, and binding partners
IMAC is particularly problematic: His-tag purification on nickel/cobalt columns can strip metal ions from metalloproteins
Cofactors don't survive purification: Heme, PLP, FAD, NAD, and metal ions often need reconstitution
Buffer composition matters enormously: Wrong pH, missing reducing agents, or incompatible additives cause irreversible damage
Activity loss is often preventable: Understanding your protein's requirements before purification is the key
The Purity-Activity Paradox
Why Do We Purify Proteins?
Purification is supposed to give us:
Homogeneous material for structural studies
Defined composition for biochemical assays
Contaminant-free samples for therapeutic development
Reproducible starting material for downstream experiments
The assumption: If we remove everything except our target protein, we'll have functional protein.
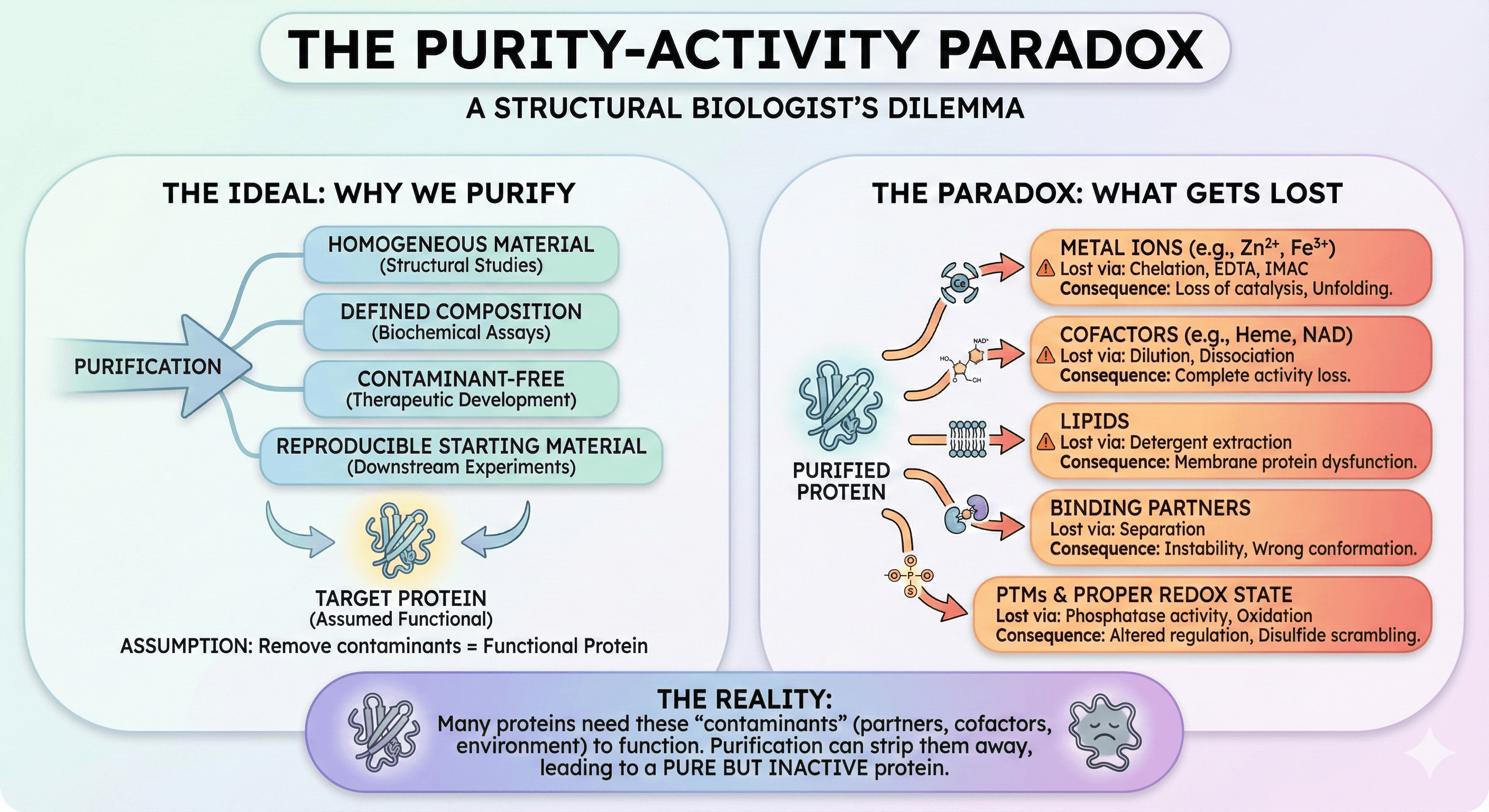
The reality: Many proteins need "contaminants" to function.
What Gets Lost
During purification, your protein can lose:
Component | How It's Lost | Consequence |
|---|---|---|
Metal ions | Chelation, EDTA, IMAC | Loss of catalysis, unfolding |
Cofactors | Dilution, dissociation | Complete activity loss |
Lipids | Detergent extraction | Membrane protein dysfunction |
Binding partners | Separation | Instability, wrong conformation |
PTMs | Phosphatase activity | Altered regulation |
Proper redox state | Oxidation | Disulfide scrambling |
The Six Ways Activity Dies During Purification
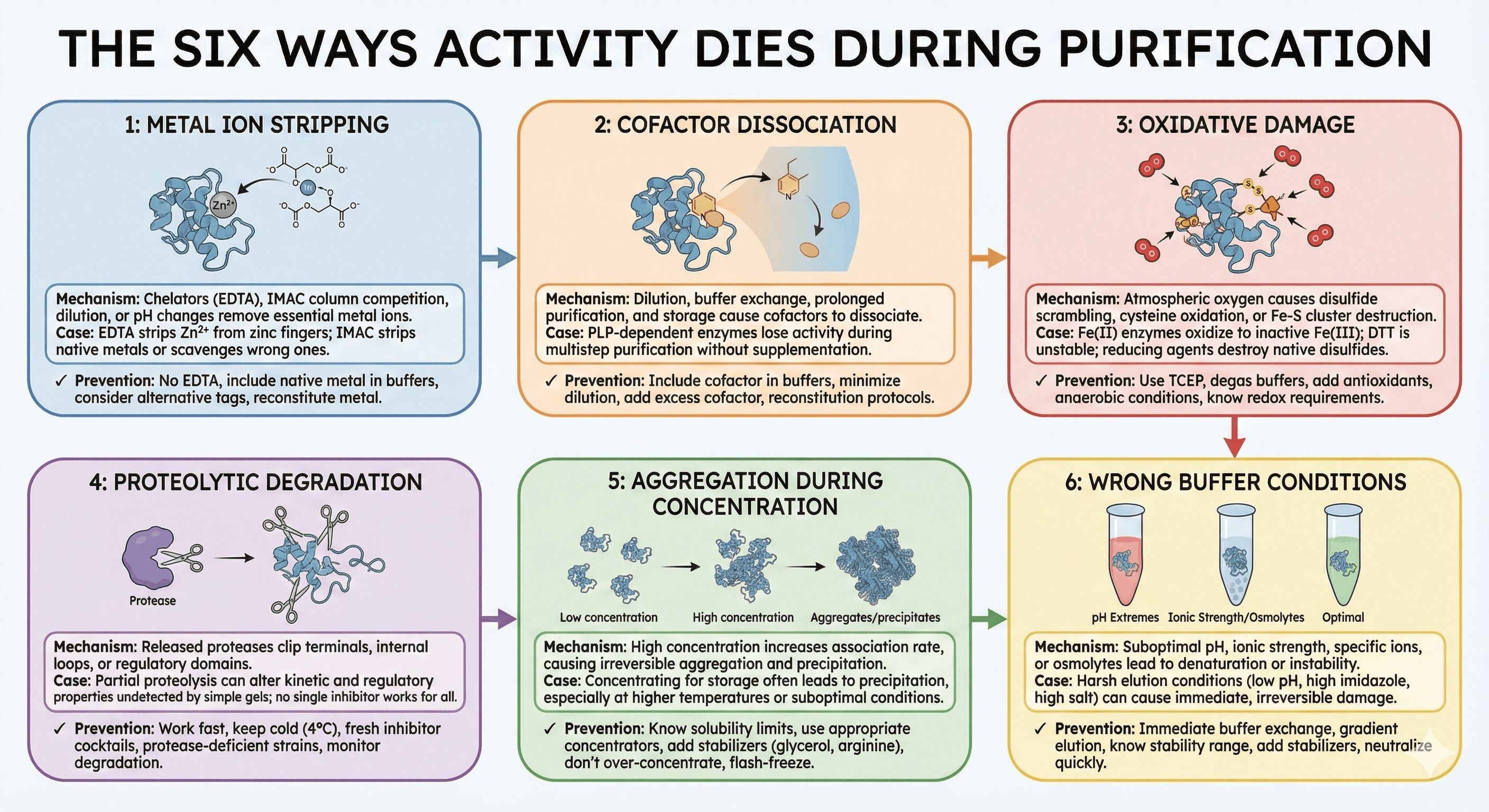
Problem 1: Metal Ion Stripping
The mechanism:
Many enzymes require metal ions for catalysis:
Zinc: Metalloproteases, carbonic anhydrase, zinc fingers
Iron: Cytochromes, iron-sulfur proteins, non-heme iron enzymes
Magnesium: Kinases, ATPases, polymerases
Copper: Oxidases, electron transport proteins
Manganese: Arginase, superoxide dismutase
During purification, metal ions are stripped by:
EDTA in buffers (even trace amounts)
Competition with IMAC column metals
Dilution below binding threshold
pH changes that alter coordination
The IMAC problem:
Research has shown that metalloproteins are particularly problematic for IMAC purification because they can scavenge metal ions from the column or have their native metals stripped. Both M-PMV and HIV-1 proteins were found to scavenge Zn²⁺ and Ni²⁺ ions from charged IMAC column matrix during elution—meaning your protein may come off the column with the wrong metal.
Case study: Zinc finger proteins
Studies on zinc-binding transcriptional regulators have demonstrated that EDTA, commonly present at 0.1-5 mM in purification buffers, can completely sequester Zn²⁺ from zinc finger proteins. The log formation constant for Zn-EDTA is 13.3, while zinc finger domains bind with constants of only 10⁷ to 10¹¹—meaning EDTA wins the competition for zinc almost every time.
Worse, some zinc-binding domains are irreversibly denatured after zinc removal and cannot refold even when zinc is added back.
Prevention:
Never use EDTA in buffers for metalloproteins
Include the native metal ion in all purification buffers
Consider alternative affinity tags (Strep-tag, FLAG) instead of His-tag
If using IMAC, reconstitute metal after elution
Problem 2: Cofactor Dissociation
The mechanism:
Many enzymes have non-covalently bound cofactors:
Heme: Cytochromes P450, peroxidases, globins
FAD/FMN: Oxidoreductases, monooxygenases
PLP (pyridoxal phosphate): Transaminases, decarboxylases
NAD/NADP: Dehydrogenases
Biotin: Carboxylases
Thiamine pyrophosphate: Decarboxylases, transketolases
Cofactors dissociate during:
Dilution (reduces effective concentration)
Buffer exchange (washes away free cofactor)
Prolonged purification (equilibrium shifts)
Storage (slow dissociation over time)
The heme problem:
Research on recombinant heme proteins has shown that production in E. coli is often limited by the host's heme biosynthesis, resulting in only partially assembled holo-heme protein. Even proteins that express with heme can lose it during purification.
In vitro reconstitution studies found that purified cytochrome c synthases contain only ~10% occupied heme; a special "heme-loading" protocol was needed to increase this to ~30%.
Case study: PLP-dependent enzymes
A transaminase purified without PLP supplementation:
Crude lysate: 100% activity (cellular PLP present)
After Ni-NTA: 60% activity (PLP diluting out)
After size exclusion: 25% activity (more dilution)
After concentration: 10% activity (most PLP lost)
The same purification with 100 μM PLP in all buffers: 95% activity retention.
Prevention:
Include cofactor in all purification buffers
Minimize dilution during purification
Add excess cofactor before storage
For heme proteins, consider reconstitution protocols
Problem 3: Oxidative Damage
The mechanism: Many proteins contain:
Free cysteines: Subject to oxidation
Iron-sulfur clusters: Oxygen-sensitive
Reduced cofactors: Air-oxidizable
Methionine residues: Oxidize to sulfoxide
Atmospheric oxygen causes:
Disulfide bond formation (aggregation, wrong structure)
Cysteine sulfenic acid formation (activity loss)
Iron-sulfur cluster destruction
Cofactor oxidation
The cysteine problem: DTT and other reducing agents are essential for proteins with free cysteines. Without them, artefactual disulfide bonds form, leading to aggregation and misfolding. However, DTT itself is unstable—its half-life is only 1.4 hours at pH 8.5 and 20°C.
For proteins that require native disulfide bonds, the situation is reversed: reducing agents will destroy the correct disulfide bonds, causing unfolding and activity loss.
Case study: Non-heme iron enzymes
Non-heme iron (II) enzymes face a particular challenge: the iron readily oxidizes to iron (III), which is catalytically inactive. A significant fraction of purified protein often contains no iron at all, requiring reconstitution after purification.
Prevention:
Use TCEP instead of DTT (more stable, compatible with IMAC)
Degas buffers for oxygen-sensitive proteins
Include antioxidants (ascorbate, reduced glutathione) where appropriate
Purify in anaerobic chamber for extreme cases
Know whether your protein needs reducing or oxidizing conditions
Problem 4: Proteolytic Degradation
The mechanism: Cell lysis releases proteases from their normal compartmentalization. These proteases can:
Clip terminal regions (removing tags or functional domains)
Cleave internal loops (fragmenting the protein)
Remove regulatory domains (altering activity)
Research on proteolysis during purification emphasizes that the problem is insidious: partial proteolysis may not be visible on a gel but can cause "striking changes to kinetic and regulatory properties."
The inhibitor challenge: No single protease inhibitor works against all proteases. Standard cocktails contain:
AEBSF/PMSF: Serine proteases
E-64: Cysteine proteases
Pepstatin: Aspartic proteases
Bestatin: Aminopeptidases
EDTA: Metalloproteases
But EDTA creates its own problems (see Problem 1), and PMSF has a half-life of only ~30 minutes in aqueous solution.
Prevention:
Work fast—minimize time between lysis and first chromatography
Keep everything cold (4°C)
Use fresh protease inhibitor cocktails
Consider protease-deficient expression strains
Monitor for degradation products on gels
Problem 5: Aggregation During Concentration
The mechanism: After purification, proteins are often concentrated for storage or downstream applications. During concentration:
Local protein concentration increases dramatically
Hydrophobic patches find each other
Aggregation-prone intermediates form
Once aggregated, protein is often irreversibly lost
Studies on protein aggregation show that the problem worsens with:
Higher target concentration
Higher temperature
Suboptimal pH
Ionic strength extremes
Surface adsorption in concentrators
The concentration trap: Protein is soluble at 1 mg/mL during purification. You concentrate to 10 mg/mL for storage. Overnight at 4°C, white precipitate appears. The protein is lost.
This happens because:
Many proteins have concentration-dependent aggregation thresholds
Concentration increases the rate of all association events, including unwanted ones
Impurities or misfolded species nucleate aggregation
Prevention:
Know your protein's solubility limit before concentrating
Use appropriate concentrator MWCO (not too close to protein MW)
Add stabilizers (glycerol, arginine, trehalose)
Don't over-concentrate—leave margin below solubility limit
Flash-freeze immediately after concentration
Problem 6: Wrong Buffer Conditions
The mechanism: Proteins have optimal conditions for stability and activity:
pH: Most enzymes have narrow pH optima
Ionic strength: Too low = aggregation; too high = denaturation
Specific ions: Some require specific cations/anions
Osmolytes: Some need stabilizers
Temperature: Some are cold-sensitive or heat-labile
During purification, conditions change multiple times: Lysis buffer → Binding buffer → Wash buffer → Elution buffer → Storage buffer. Each transition is an opportunity for damage.
Case study: The elution step
Many elution conditions are harsh:
Low pH (4.0) for Protein A
High imidazole (500 mM) for IMAC
High salt for ion exchange
Organic solvents for hydrophobic interaction
The protein may not survive these conditions, even briefly.
Prevention:
Buffer exchange immediately after elution
Use gradient elution where possible (less harsh)
Know your protein's pH stability range
Include stabilizers in elution buffers
Neutralize immediately after low-pH elution
The Diagnostic Workflow
When Activity Is Lost, Ask These Questions
Step 1: At which step did activity disappear?
Run activity assays at each purification stage:
Lysate
Post-affinity
Post-ion exchange
Post-gel filtration
After concentration
After storage
The step where activity drops is where the damage occurs.
Step 2: What changed at that step?
Step | Common Culprits |
|---|---|
Lysis | Proteolysis, oxidation, metal loss |
Affinity (IMAC) | Metal stripping, wrong metal incorporation |
Ion exchange | pH stress, ionic strength shock |
Gel filtration | Dilution of cofactors, partner loss |
Concentration | Aggregation, surface adsorption |
Storage | Oxidation, freeze-thaw damage |
Step 3: Can activity be rescued?
Try reconstitution:
Add back suspected metal ion
Add cofactor in excess
Add reducing agent
Add suspected binding partner
Change to optimal buffer
If activity is rescued, you've identified the problem.
Prevention Strategies
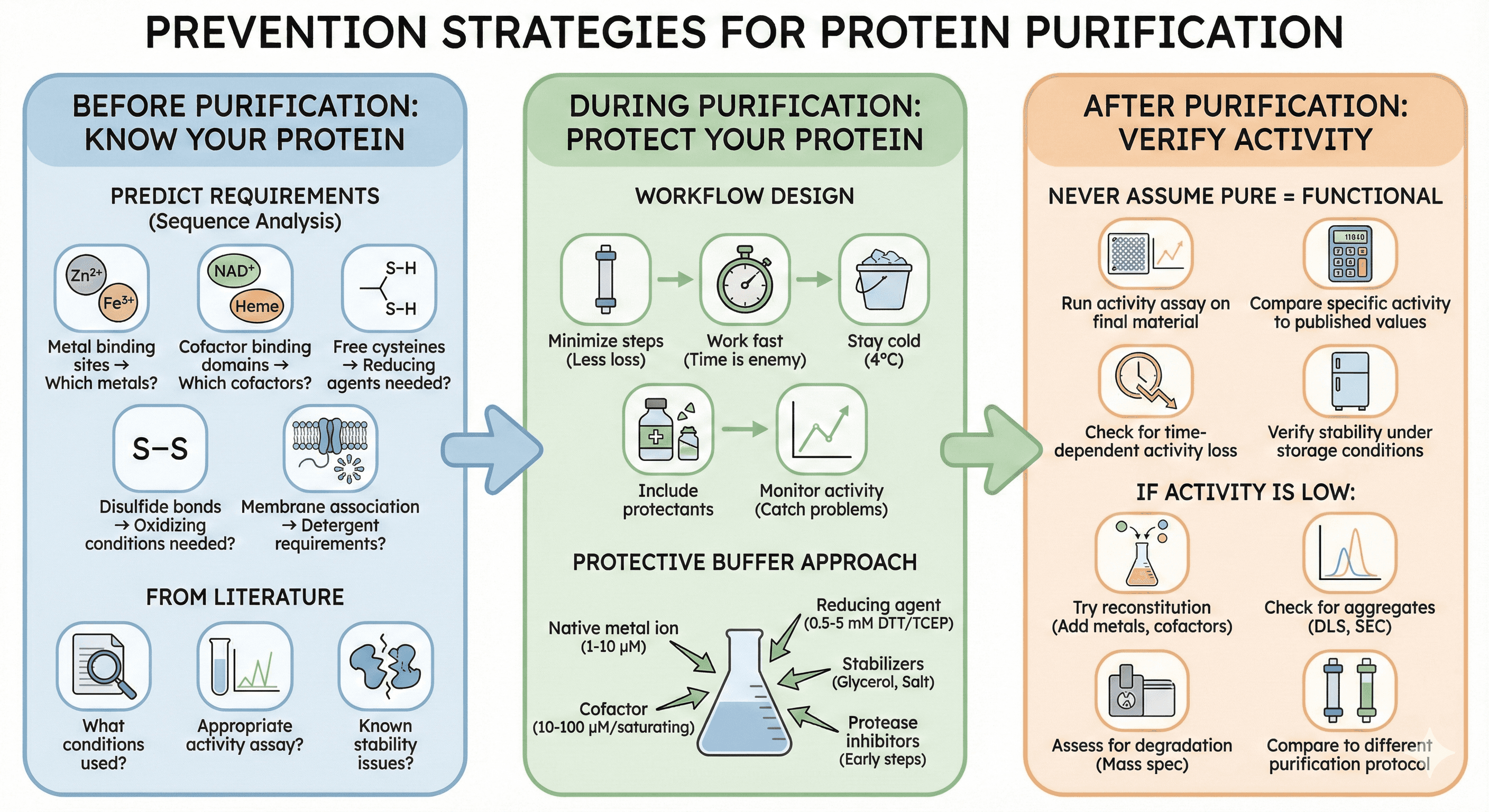
Before Purification: Know Your Protein
Predict requirements: From sequence analysis:
Metal binding sites → Which metals?
Cofactor binding domains → Which cofactors?
Free cysteines → Reducing agents needed?
Disulfide bonds → Oxidizing conditions needed?
Membrane association → Detergent requirements?
From literature:
What conditions have others used?
What activity assay is appropriate?
What are known stability issues?
During Purification: Protect Your Protein
Design the workflow with function in mind:
Minimize steps: Every column is an opportunity for loss
Work fast: Time is the enemy for unstable proteins
Stay cold: 4°C slows most degradation processes
Include protectants: Metals, cofactors, reducing agents
Monitor activity: Not just purity—catch problems early
The protective buffer approach: Instead of standard purification buffers, design buffers that include:
The native metal ion (1-10 μM range)
The cofactor (10-100 μM range, or saturating)
Appropriate reducing agent (0.5-5 mM DTT or TCEP)
Stabilizers (5-10% glycerol, 100-500 mM salt)
Protease inhibitors (during early steps)
After Purification: Verify Activity
Never assume pure means functional:
Run activity assay on final material
Compare specific activity to published values
Check for time-dependent activity loss
Verify activity is stable under storage conditions
If activity is low:
Try reconstitution (add metals, cofactors)
Check for aggregates (DLS, SEC)
Assess for degradation (mass spec)
Compare to different purification protocol
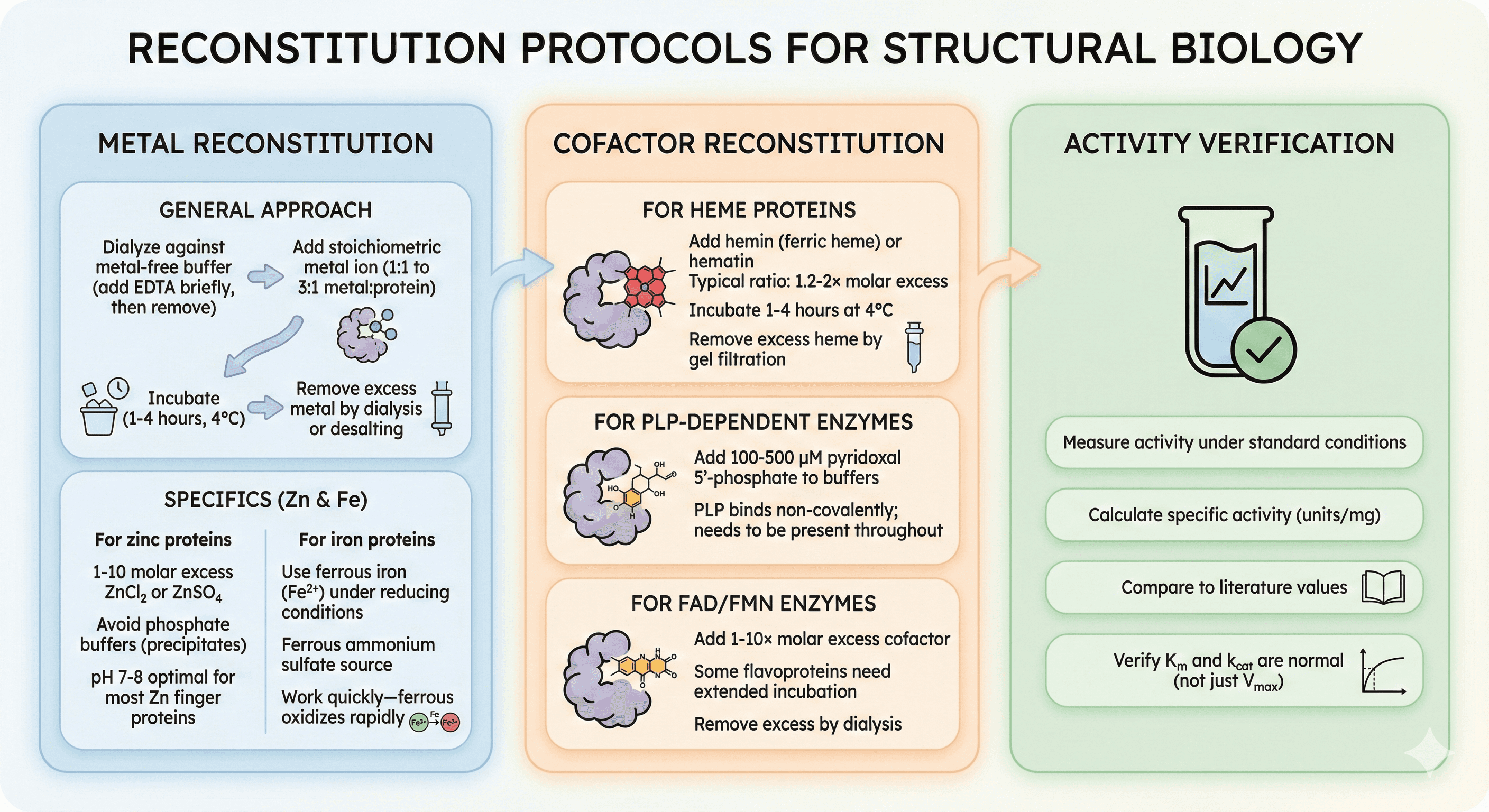
Reconstitution Protocols
Metal Reconstitution
General approach:
Dialyze protein against metal-free buffer (add EDTA briefly, then remove)
Add stoichiometric metal ion (1:1 to 3:1 metal:protein)
Incubate (1-4 hours, 4°C)
Remove excess metal by dialysis or desalting
For zinc proteins:
1-10 molar excess ZnCl₂ or ZnSO₄
Avoid phosphate buffers (zinc phosphate precipitates)
pH 7-8 optimal for most zinc finger proteins
For iron proteins:
Use ferrous iron (Fe²⁺) under reducing conditions
Ferrous ammonium sulfate is common source
Work quickly—ferrous oxidizes rapidly
Cofactor Reconstitution
For heme proteins:
Add hemin (ferric heme) or hematin
Typical ratio: 1.2-2× molar excess
Incubate 1-4 hours at 4°C
Remove excess heme by gel filtration
For PLP-dependent enzymes:
Add 100-500 μM pyridoxal 5'-phosphate to buffers
PLP binds non-covalently; needs to be present throughout
For FAD/FMN enzymes:
Add 1-10× molar excess cofactor
Some flavoproteins need extended incubation
Remove excess by dialysis
Activity Verification
After reconstitution:
Measure activity under standard conditions
Calculate specific activity (units/mg)
Compare to literature values
Verify Km and kcat are normal (not just Vmax)
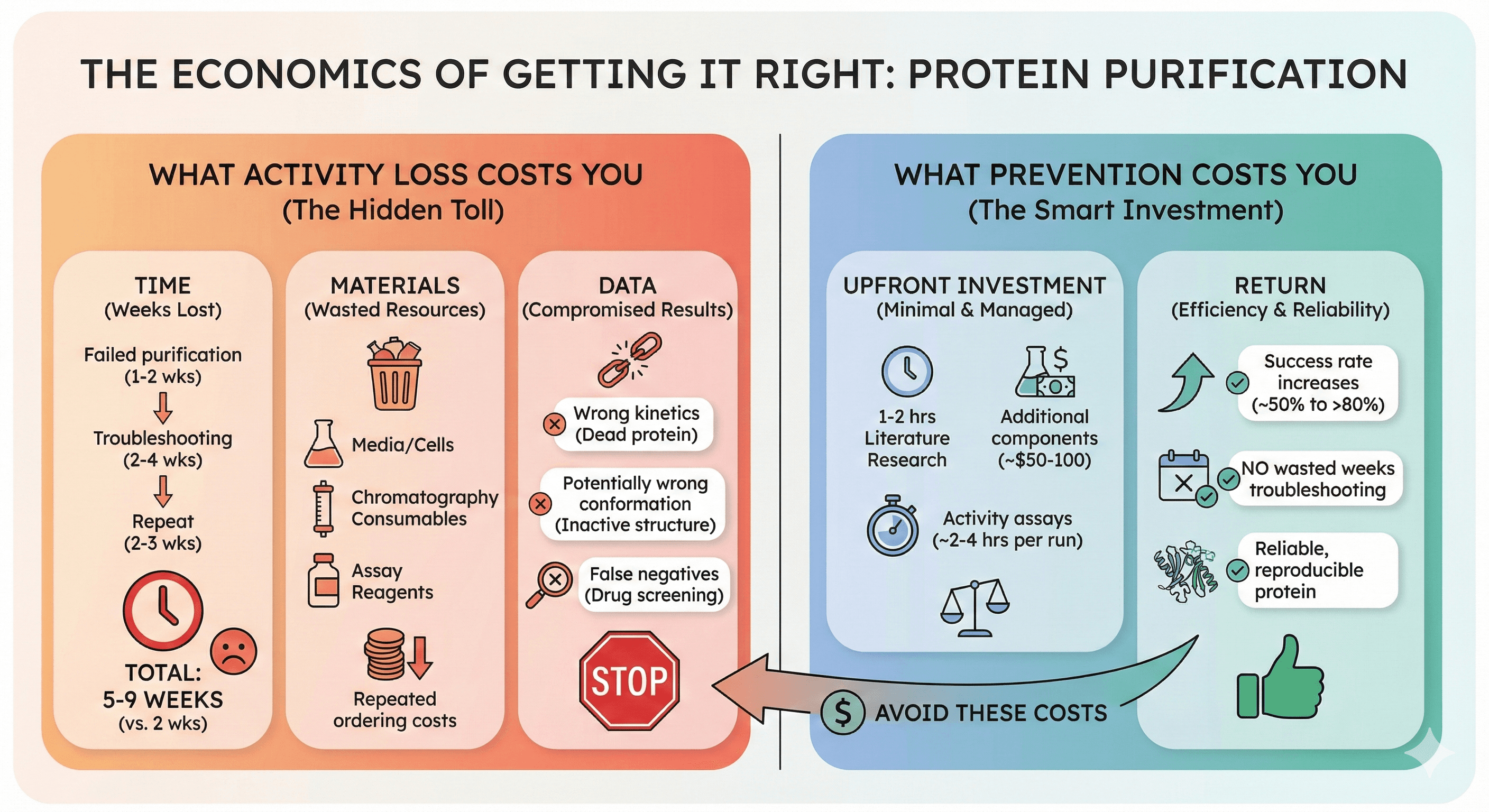
The Economics of Getting It Right
What Activity Loss Costs You
Time:
Failed purification: 1-2 weeks
Troubleshooting: 2-4 weeks
Repeat with modifications: 2-3 weeks
Total: 5-9 weeks for what should have been 2 weeks
Materials:
Wasted expression (media, cells, inducer)
Wasted chromatography consumables
Wasted activity assay reagents
Repeated ordering costs
Data:
Biochemical characterization with dead protein = wrong kinetics
Structural studies with inactive protein = potentially wrong conformation
Drug screening with compromised target = false negatives
What Prevention Costs You
Upfront investment:
1-2 hours literature research on protein requirements
Additional buffer components (~$50-100)
Activity assays at each step (~2-4 hours per purification)
Return:
First-time success rate improves from ~50% to >80%
No wasted weeks troubleshooting
Reliable, reproducible protein
The Bottom Line
Purity is not activity. The most common reasons for activity loss during purification are:
Problem | Solution |
|---|---|
Metal stripping | Avoid EDTA, include native metal |
Cofactor loss | Include cofactor in all buffers |
Oxidative damage | Use appropriate reducing agents |
Proteolysis | Work fast, use inhibitors |
Aggregation | Know solubility limits, use stabilizers |
Wrong conditions | Optimize buffer for your specific protein |
The difference between a failed purification and a successful one is often not technique—it's knowledge. Knowing what your protein needs before you start purification prevents most activity loss problems.
Protein-Specific Purification Planning
For researchers working with challenging proteins, platforms like Orbion can help identify potential purification problems before you encounter them:
Metal binding site prediction: Know which metals your protein requires
Cofactor binding analysis: Identify cofactor requirements from sequence
PTM prediction: Understand modification requirements
Disorder and aggregation prediction: Anticipate concentration limits
The goal is to design your purification around your protein's requirements—not discover them after activity is already lost.
References
Block H, et al. (2009). Immobilized-metal affinity chromatography (IMAC): a review. Methods in Enzymology, 463:439-473. PMC3134162
Krizek BA, et al. (1993). That zincing feeling: the effects of EDTA on the behaviour of zinc-binding transcriptional regulators. Journal of Biological Chemistry, 268(17):12387-12392. PMC1133908
Londer YY, et al. (2018). Improved method for the incorporation of heme cofactors into recombinant proteins using Escherichia coli Nissle 1917. Biochemistry, 57(19):2764-2770. Link
Sutherland MC, et al. (2021). In vitro reconstitution reveals major differences between human and bacterial cytochrome c synthases. eLife, 10:e64891. PMC8112865
Ocaña-Calahorro F, et al. (2022). Protein purification strategies must consider downstream applications and individual biological characteristics. Protein Expression and Purification, 191:106026. PMC8991485
Ryan BJ & Henehan GT. (2016). Avoiding proteolysis during protein purification. Methods in Molecular Biology, 1485:53-69. PubMed
Bondos SE & Bhattacharya A. (2003). Detection and prevention of protein aggregation before, during, and after purification. Analytical Biochemistry, 316(2):223-231. PubMed
Cleland WW. (1964). Dithiothreitol, a new protective reagent for SH groups. Biochemistry, 3:480-482. ScienceDirect
Book a 20-Minute Demo
Sign up free for unlimited Overview runs — summary, sequence-based analysis, homology search. For the full Characterization — PTMs, binding sites, stability variants, construct design — book a demo and we'll run your target live.
