Blog
Orbion Team
AlphaFold's Limitations: What It Can't Predict (And How to Fill the Gaps)

AlphaFold solved protein folding. But structure prediction is just the beginning. When you're trying to actually express, purify, crystallize, or drug a protein—a static structure is only the starting point. Here's what AlphaFold can't tell you, and how modern tools are filling the gaps.
Key Takeaways
AlphaFold predicts structure, not context: No PTMs, binding sites, or membrane topology
Biased toward Apo states: Struggles with drug-bound (Holo) conformations and conformational ensembles
pLDDT ≠ stability: High-confidence predictions can still aggregate in the lab
Missing quaternary structure: Original AF2 was blind to protein-protein interactions
The solution: AlphaFold Multimer, MSA subsampling, and AI context layers (like Orbion)
The Gap Between Structure and Success
When DeepMind released AlphaFold2 in 2020, it felt seismic. For the first time, we could predict protein structures from sequence with near-experimental accuracy—in minutes, not months. It solved a 50-year grand challenge.
But if you talk to structural biologists and drug hunters today, the excitement is paired with pragmatism. AlphaFold is a brilliant architect, but it's also a rigid one. It gives you a snapshot, not the full movie. And when you're trying to actually work with a protein in the lab, that snapshot is just the beginning.
Let me break down the six critical gaps—and how we're fixing them.
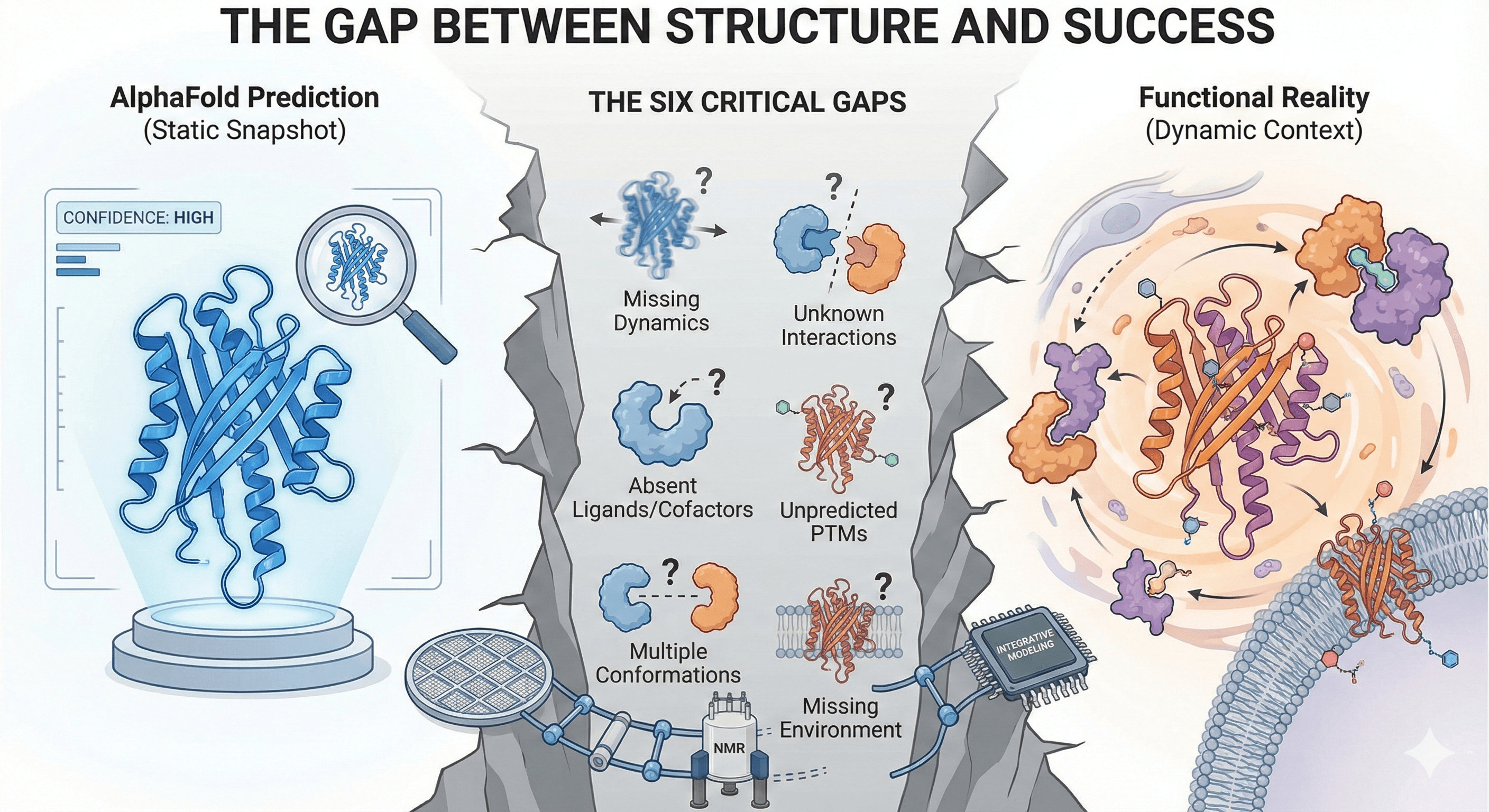
The Six Critical Gaps in AlphaFold Predictions
1. Post-Translational Modifications (PTMs): The Missing Context
AlphaFold predicts your protein as a naked polypeptide chain. But in reality, proteins are heavily decorated after translation. Glycosylation, phosphorylation, acetylation, ubiquitination, disulfide bonds—these modifications don't just decorate your protein, they fundamentally change its behavior.
Why this matters:
A glycosylation site can determine whether your protein folds correctly in mammalian cells
A phosphorylation site might be essential for function—miss it, and your construct is useless
A disulfide bond you didn't account for means your purified protein is a misfolded aggregate
The numbers:
~5% of human proteome is glycosylated at some point
Average eukaryotic protein has 3-5 phosphorylation sites
Therapeutic antibodies require specific N-glycosylation at Asn297 for effector function
AlphaFold won't tell you any of this. You discover it the hard way when your expression fails.
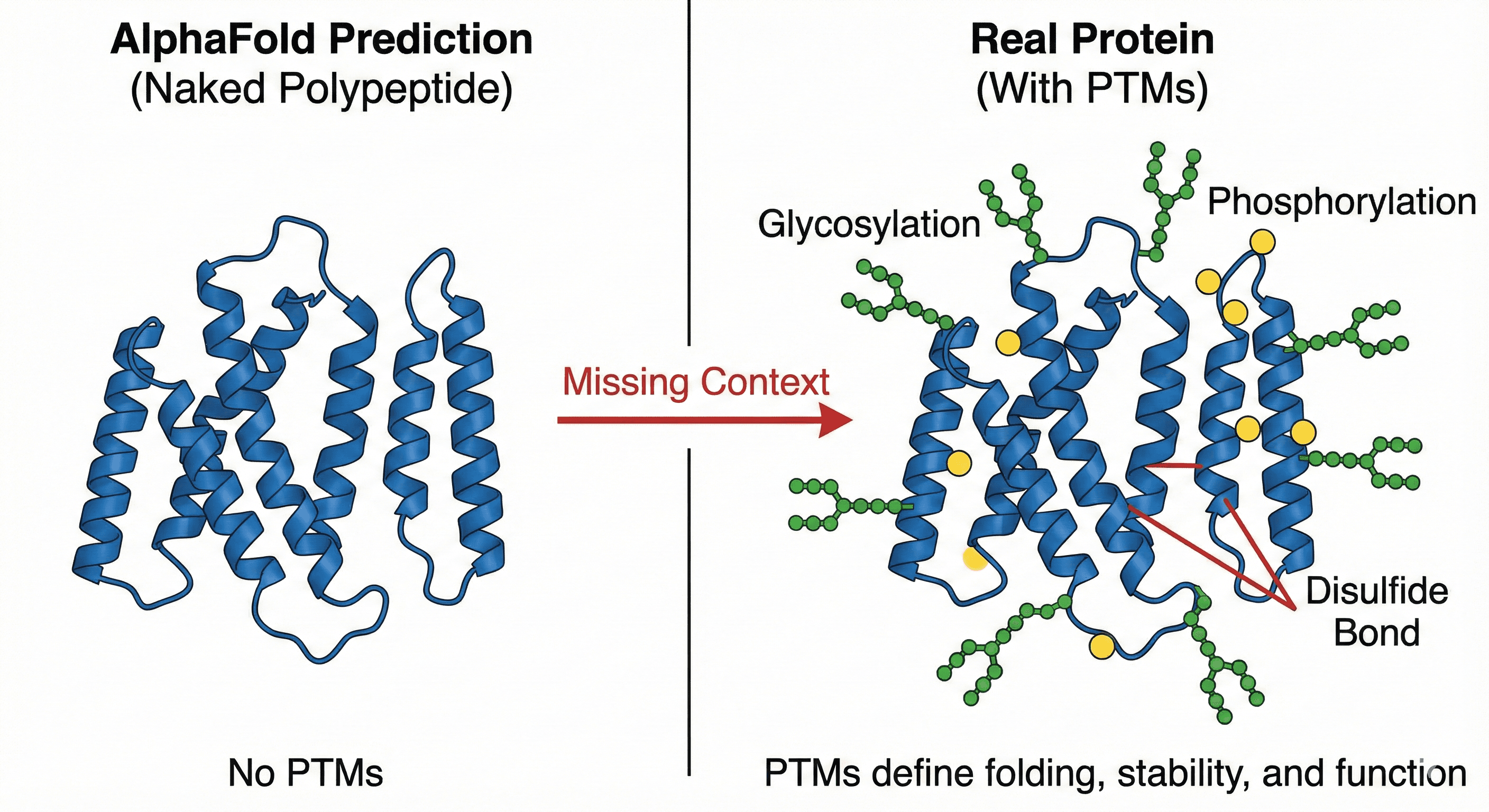
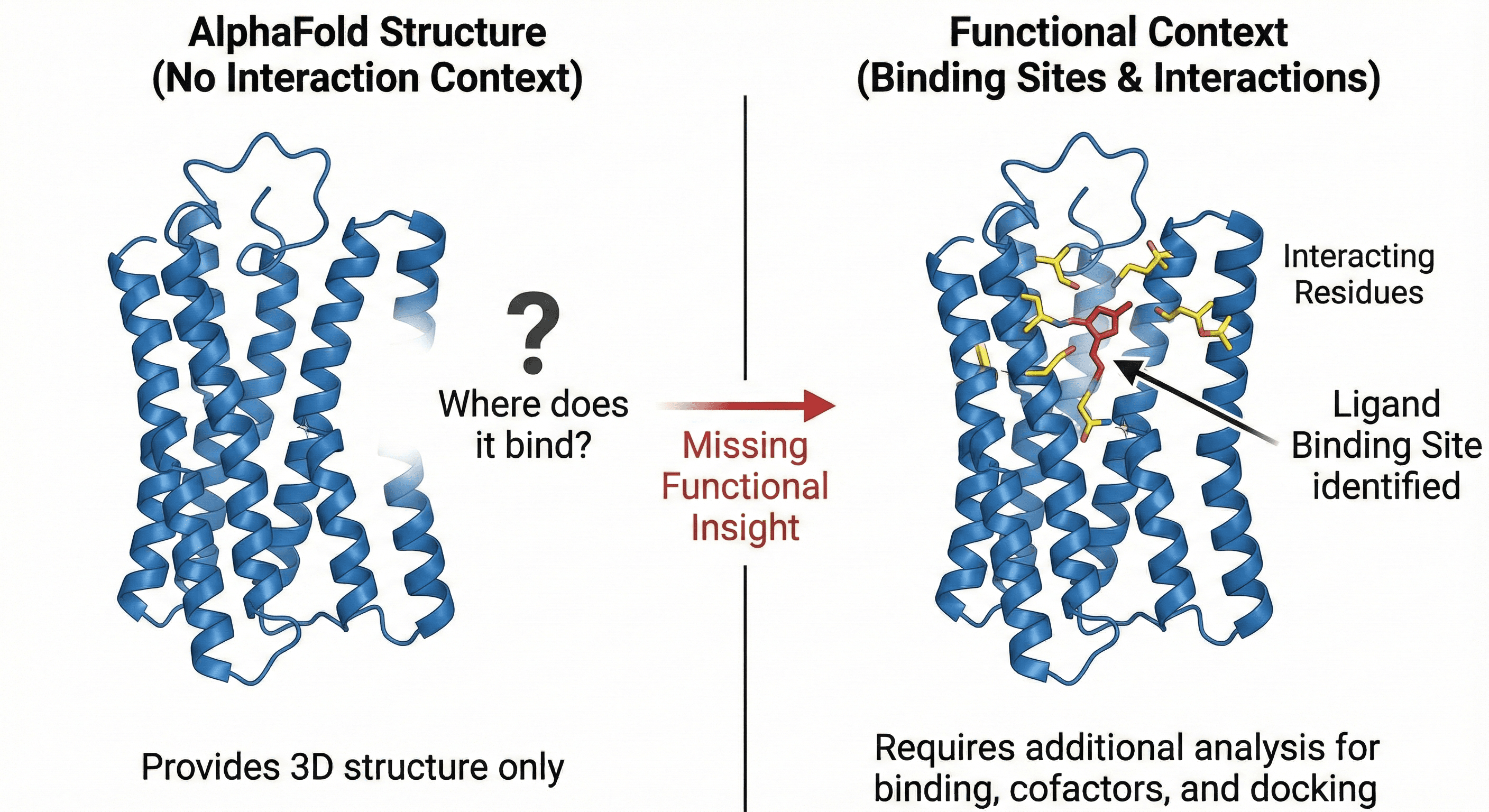
2. Binding Sites and Molecular Interactions: Where Does It Act?
AlphaFold gives you a structure, but it doesn't tell you what that structure does. Where does your protein bind its substrate? Which residues coordinate the metal cofactor? Where does the drug molecule dock?
The problem:
You can run docking simulations on an AlphaFold structure, but identifying the binding sites, understanding the interaction interfaces, knowing which residues are critical—that requires additional analysis
For drug discovery and protein engineering, this is often the most important information
And it's completely absent from an AlphaFold prediction
Example: You're designing a GPCR inhibitor. AlphaFold gives you the 7-transmembrane bundle. But which residues in the orthosteric pocket interact with your ligand? Which allosteric sites could you target? You need binding site prediction—not just structure.
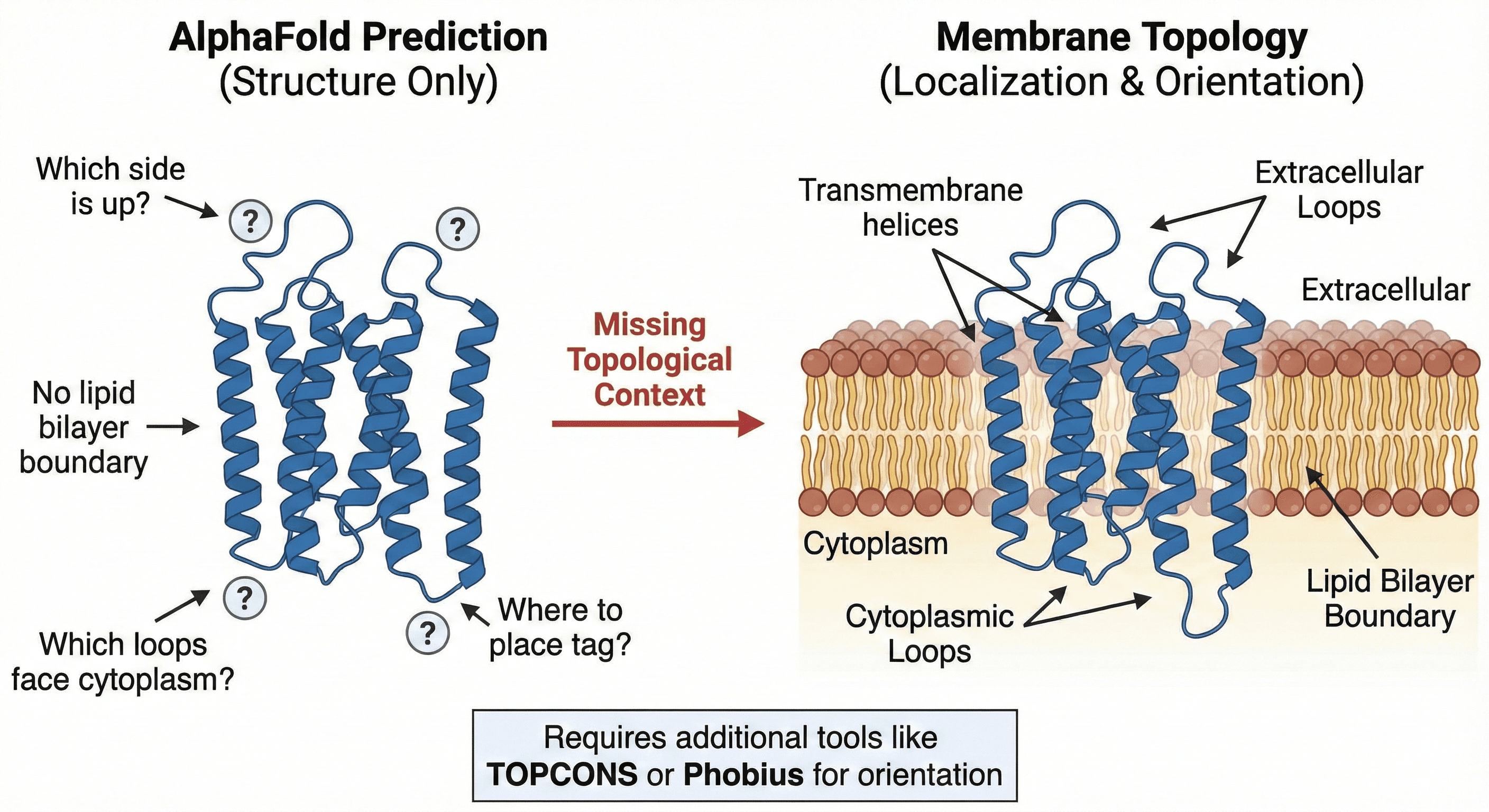
3. Membrane Topology and Localization: Which Side Is Up?
If you're working with a membrane protein—and roughly 30% of all proteins are membrane-associated—AlphaFold's prediction becomes even more incomplete.
What's missing:
Which regions are transmembrane?
Which loops face the cytoplasm versus the extracellular space?
Where should you place your purification tag to avoid disrupting function?
What's the lipid bilayer boundary?
AlphaFold predicts structure, not topology. For GPCRs, ion channels, and transporters, this gap is significant. You need to know the orientation of your protein in the membrane before you can design constructs that will actually express and purify.
Technical note: Tools like TOPCONS and Phobius can predict topology from sequence, but integrating this with AlphaFold's structure requires manual work—or automated context layers.
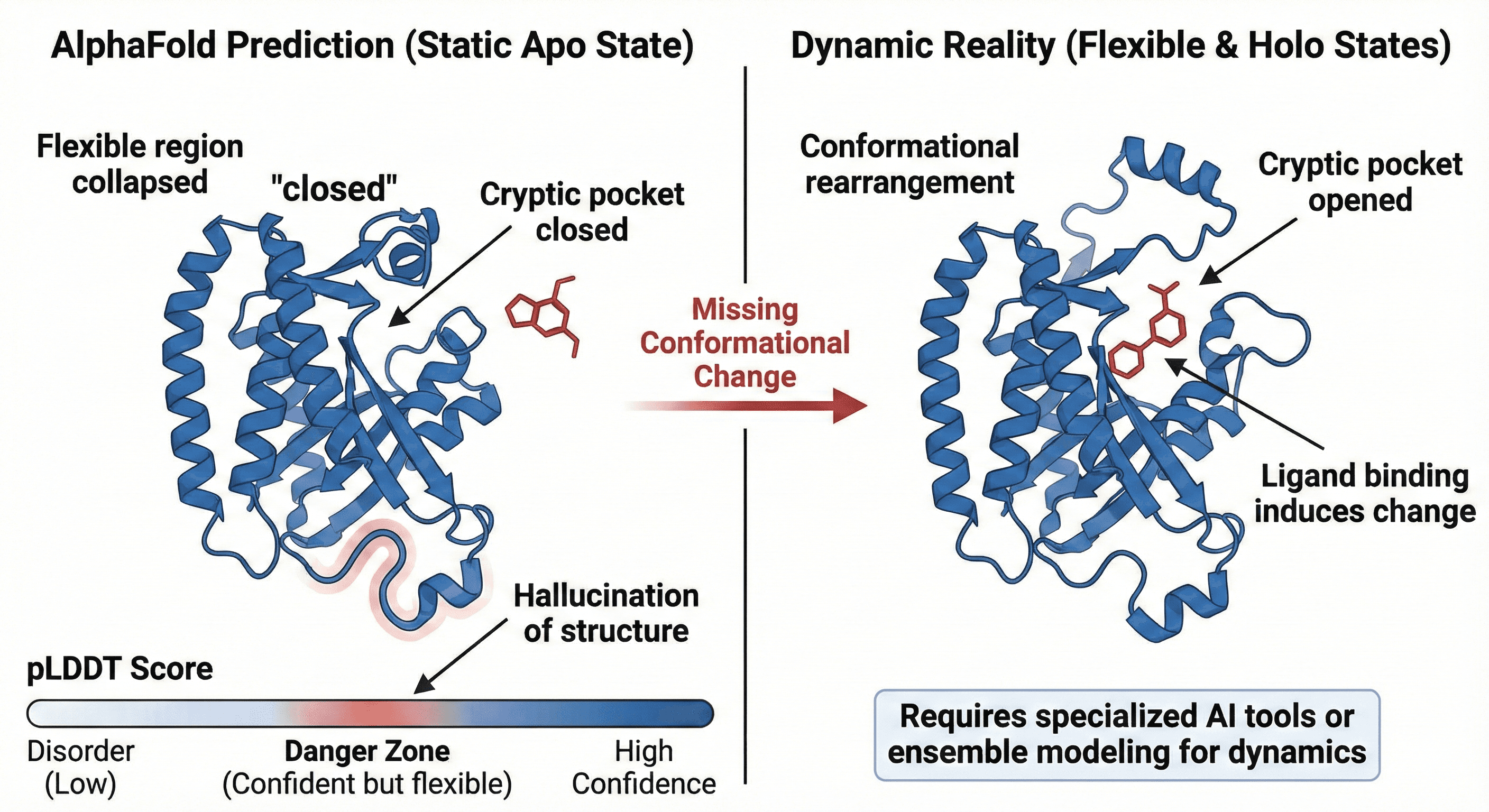
4. The Apo vs. Holo Problem: Static Structure, Dynamic Reality
This is where AlphaFold hits a fundamental biophysical wall.
The training bias: AlphaFold learned from the PDB (Protein Data Bank), which is dominated by crystal structures. Crystal structures are often frozen in the most stable, lowest-energy conformation—the Apo (ligand-free) state.
The problem for drug discovery: When a ligand binds (creating the Holo state), proteins often undergo significant conformational rearrangements:
Loop movements
Domain closures
Cryptic pocket opening
AlphaFold tends to collapse flexible regions into the lowest-energy Apo state. If you attempt structure-based drug design (SBDD) on a standard AF2 model, you often find the cryptic pockets are "closed," rendering docking simulations useless.
The pLDDT trap: We're trained to trust the pLDDT (per-residue confidence) score:
Deep blue (pLDDT > 90) = high confidence
Orange/red (pLDDT < 50) = probably disorder
But AF2 often correctly predicts intrinsic disorder as low confidence. The danger lies in the middle ground: regions where AF2 is "confident" about a structure that, in reality, is highly flexible or transient. It can hallucinate a structured loop where a flexible tether exists.
Example: An enzyme in its open conformation vs. substrate-bound closed state. AlphaFold gives you the open state. The drug binding site? Closed off in the prediction.
5. Stability and Expression Feasibility: Will It Actually Fold?
This is perhaps the biggest gap for experimental biologists.
AlphaFold predicts what a protein might look like if it folds correctly. It doesn't tell you whether it will fold correctly in your expression system.
The disconnect:
pLDDT measures prediction confidence, not protein stability
A protein can have a beautiful, high-confidence AlphaFold structure and still aggregate the moment you try to express it
The structure might be thermodynamically favorable but kinetically inaccessible
What AlphaFold doesn't tell you:
Which mutations would stabilize your protein (thermostabilizing mutations)
Whether your construct will express in E. coli or requires mammalian cells
Which regions are aggregation hotspots
How to rescue a protein that's failing in the lab
For challenging proteins—GPCRs, aggregation-prone targets, intrinsically unstable domains—this is where projects fail. Not because we don't know the structure, but because we can't make the protein.
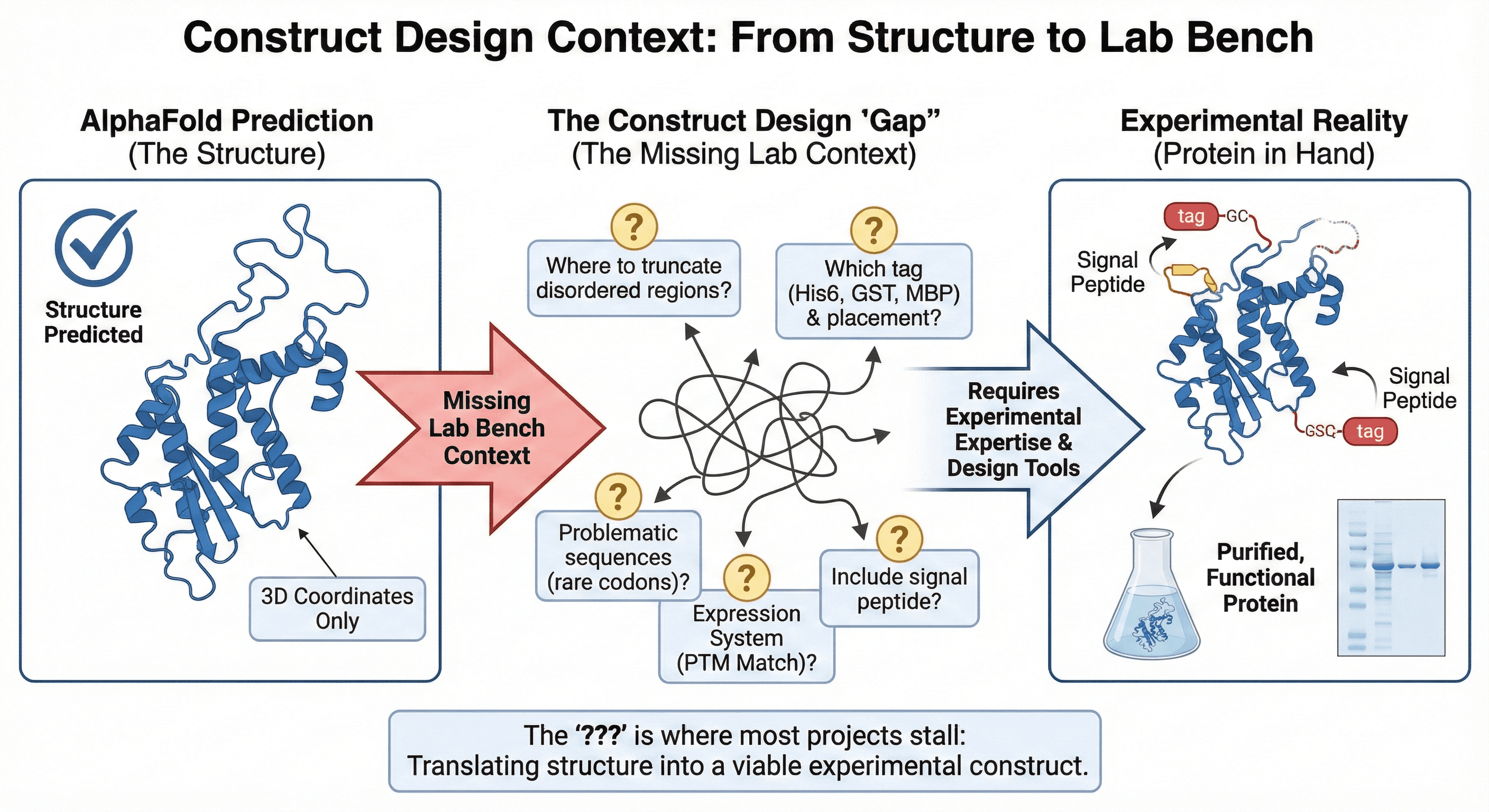
6. Construct Design Context: From Structure to Lab Bench
When you're designing a construct for expression, you need to make dozens of decisions:
Where to truncate disordered regions?
Which tag to use (His6, GST, MBP), and where to place it?
Should you include a signal peptide?
Which expression system matches your protein's PTM requirements?
Are there problematic sequences that will cause expression issues (rare codons, internal start sites)?
AlphaFold doesn't help with any of this. It gives you the structure; you're on your own for everything else.
Example workflow gap:
AlphaFold predicts structure ✅
You need to express it...
???
Protein in hand (hopefully)
The "???" is where most projects stall.
The Modern Toolkit: How We're Filling the Gaps
Science abhors a vacuum. Where AlphaFold left biophysical gaps, the community has rushed in. Here's how we're turning static predictions into dynamic, usable data.
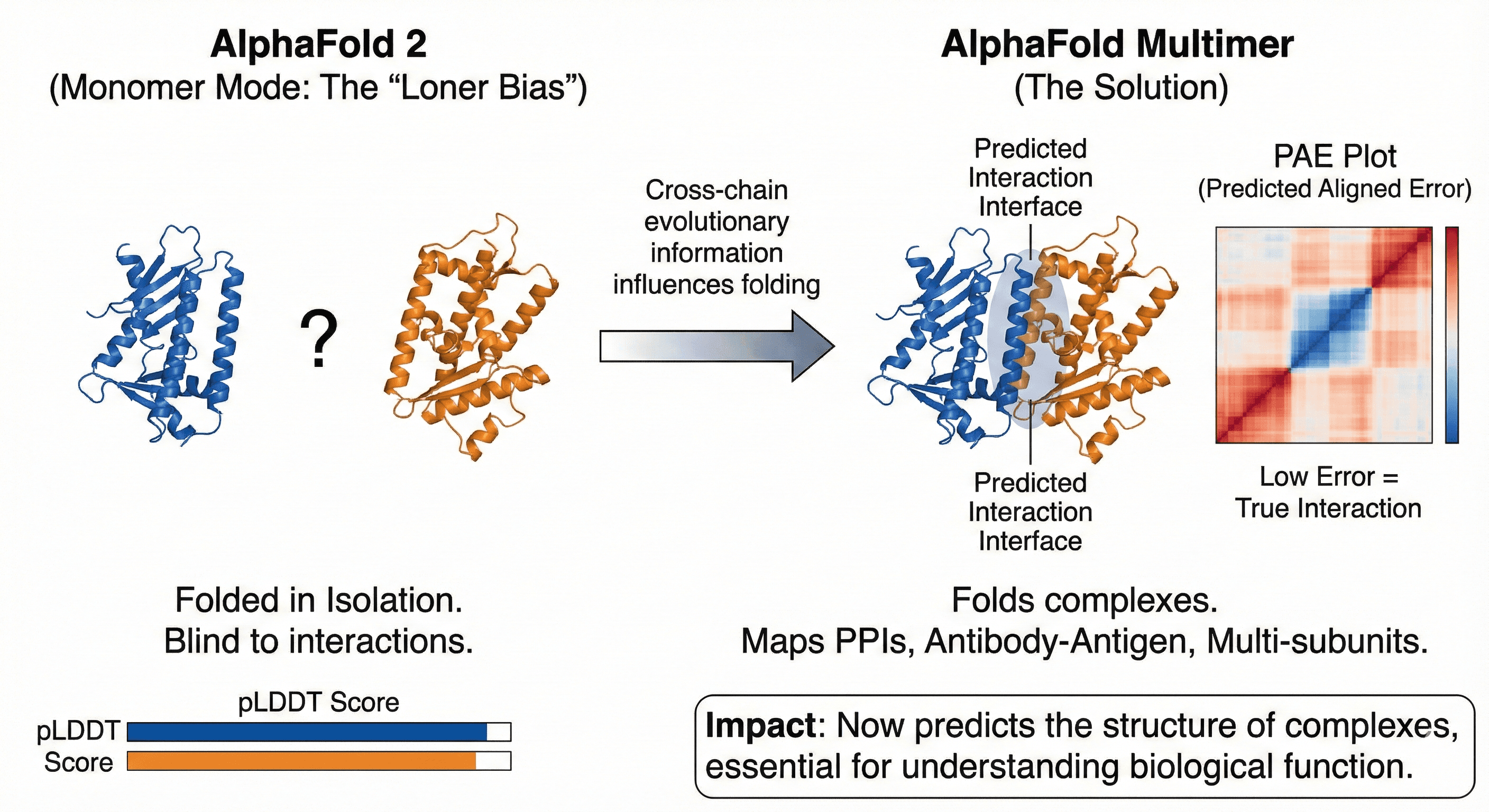
1. Solving Quaternary Structure: AlphaFold Multimer
To address the "loner bias" (AF2's blindness to protein-protein interactions), DeepMind released AlphaFold Multimer.
The mechanism: Rather than folding chains in isolation, it allows cross-chain evolutionary information to influence the folding path.
The use case:
Critical for mapping Protein-Protein Interactions (PPIs)
G-protein coupling to GPCRs
Antibody-antigen interfaces
Multi-subunit complexes
The metric: PAE (Predicted Aligned Error) plots show whether two proteins actually interact or are just floating near each other in space.
Impact: Now you can predict the structure of complexes, not just monomers. Essential for understanding biological function.
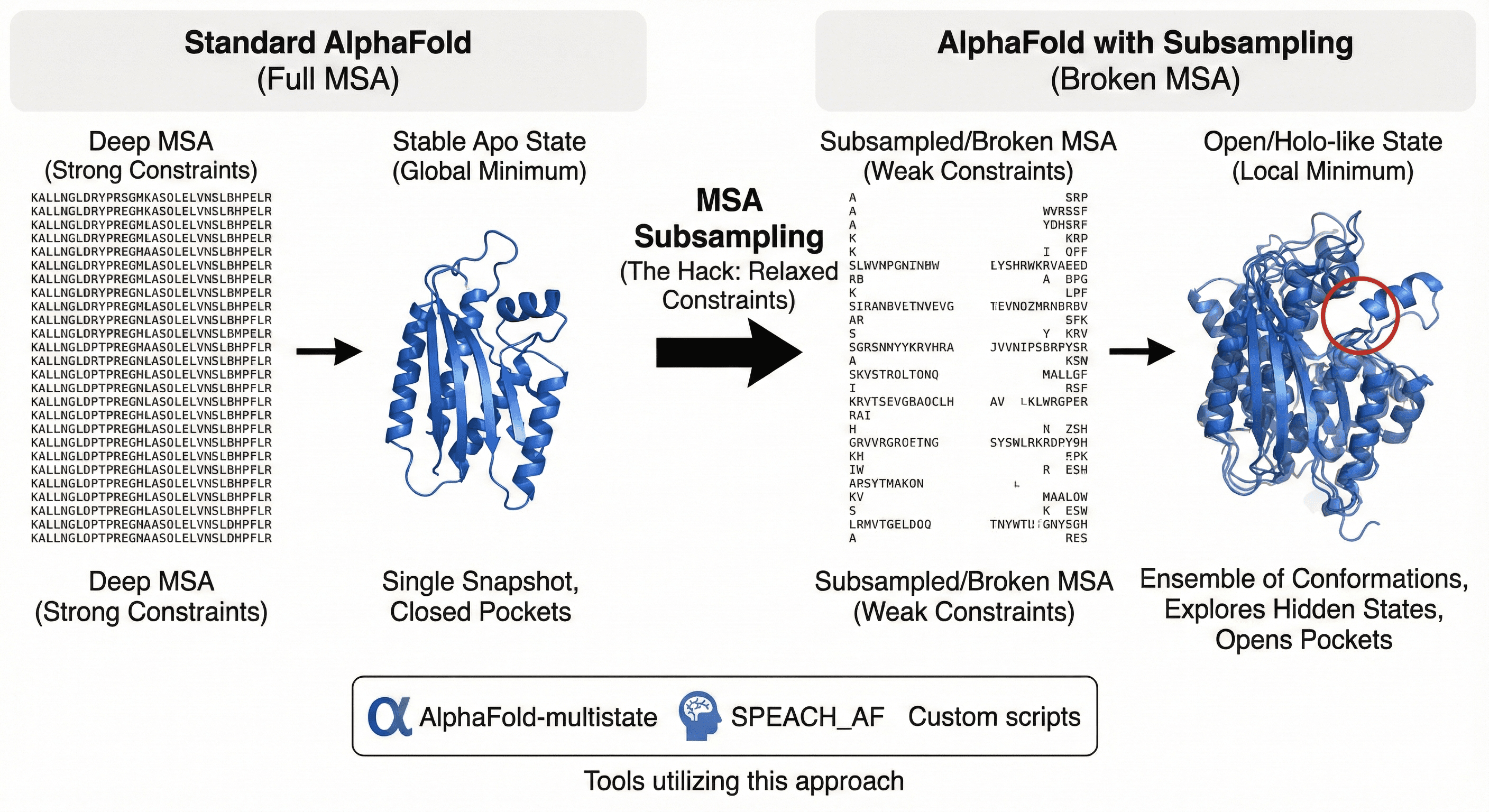
2. Sampling Hidden States: MSA Subsampling
If AF2 gives us the "global minimum" (stable Apo state), how do we find the local minima (active/excited/Holo states)?
The hack: MSA Subsampling
AlphaFold relies on deep Multiple Sequence Alignments (MSAs) to constrain protein structure. By intentionally "breaking" this data—randomly dropping sequences or reducing cluster depth—researchers can relax these evolutionary constraints.
The result: This forces the AI to explore alternative conformations, effectively turning a snapshot into a movie.
Tools that use this:
AlphaFold-multistate
SPEACH_AF
Custom MSA dropout scripts
This is how we begin to see the "open" pockets required for drug binding that are closed in the standard Apo prediction.
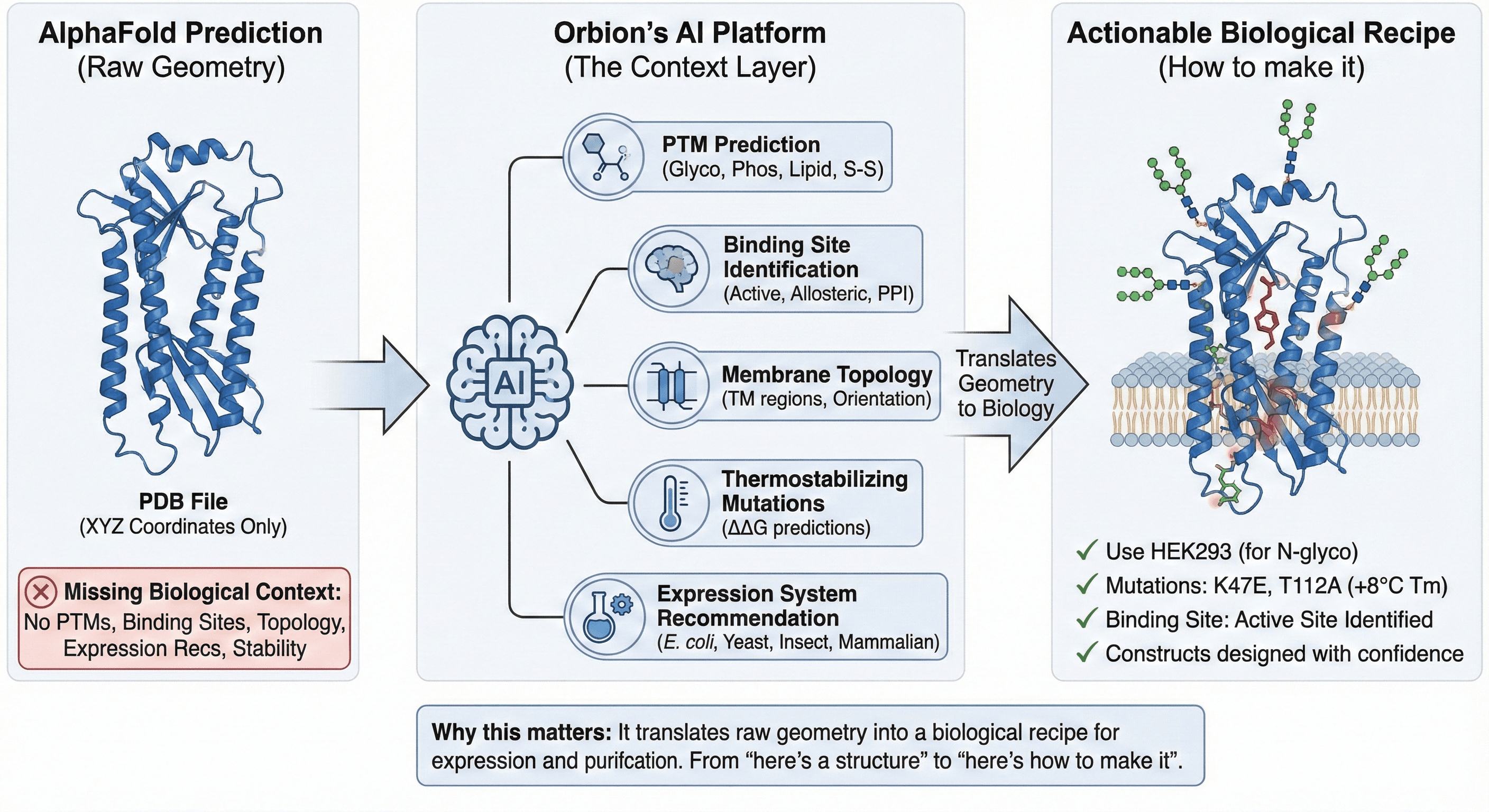
3. The Context Layer: Orbion's AI Platform
A PDB file is just coordinates. It lacks the biological context required for wet-lab experimentation.
The gap AlphaFold leaves:
No PTM predictions
No binding site identification
No membrane topology annotation
No expression system recommendations
No stability optimization
The solution: Platforms like Orbion act as a post-processing intelligence layer. They analyze the AlphaFold structure to provide:
✓ PTM prediction - N/O-glycosylation, phosphorylation, lipidation, disulfide bonds
✓ Binding site identification - Active sites, allosteric sites, protein-protein interfaces
✓ Membrane topology - Transmembrane regions, signal peptides, orientation
✓ Thermostabilizing mutations - ΔΔG predictions for stability engineering
✓ Expression system recommendation - E. coli vs yeast vs insect vs mammalian based on PTM requirements
Why this matters: It translates raw geometry into a biological recipe for expression and purification. You go from "here's a structure" to "here's how to make it."
Example workflow:
AlphaFold predicts structure
Orbion analyzes structure for PTMs, topology, binding sites
Orbion recommends: "This protein requires N-glycosylation → use HEK293 cells"
Orbion suggests stabilizing mutations: "K47E and T112A increase Tm by 8°C"
You design constructs with confidence
The Modern Structural Biology Stack
For the practicing biologist, here's how these tools fit together:
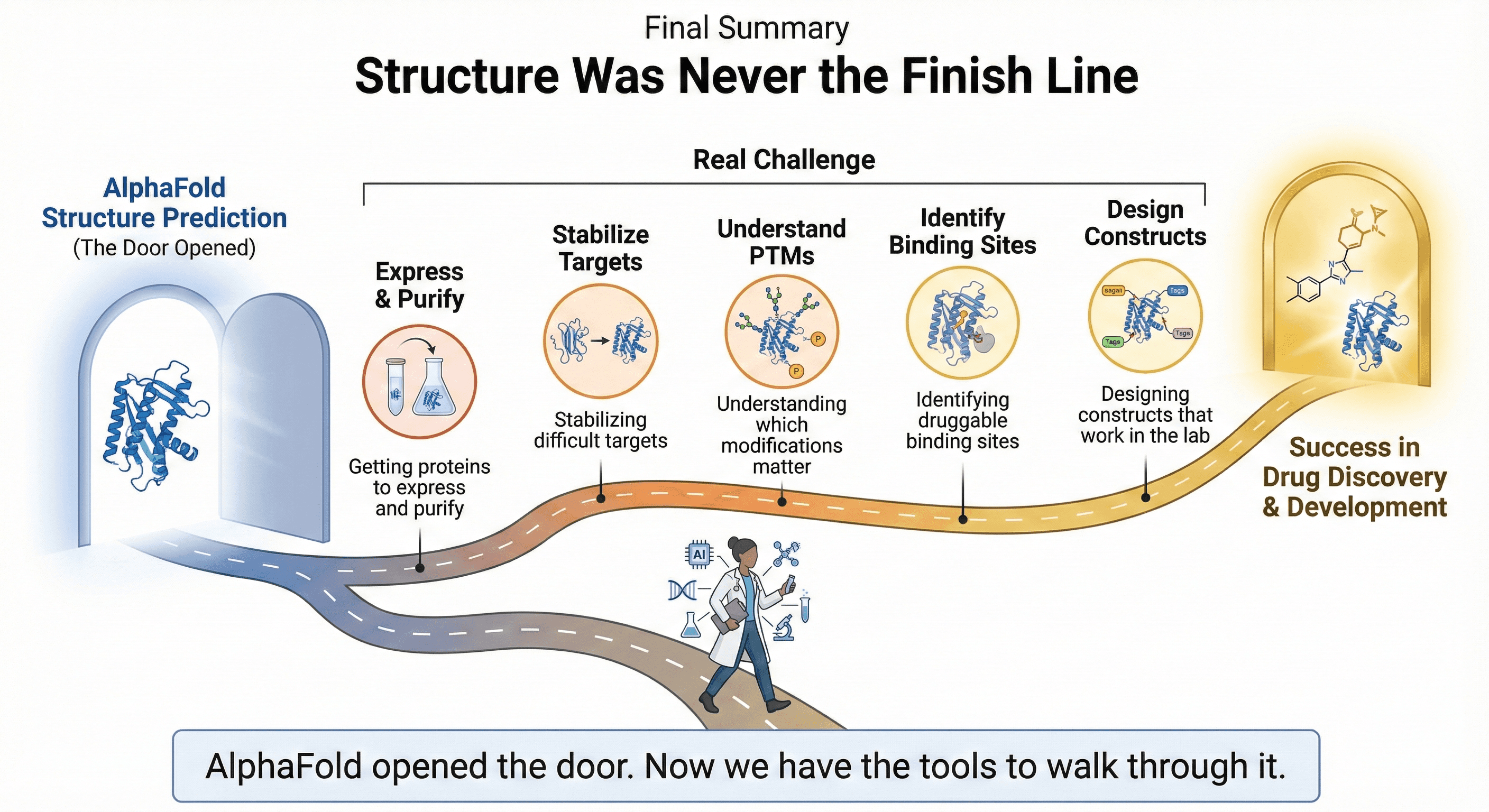
Structure Was Never the Finish Line
AlphaFold is remarkable. It solved a 50-year grand challenge. Every structural biologist uses it, and it has accelerated research across the field.
But the protein folding problem was never the only problem. For those working in drug discovery, protein engineering, and therapeutic development, structure prediction was always just one step in a longer journey.
The real challenges remain:
Getting proteins to express and purify
Stabilizing difficult targets for structural studies
Understanding which modifications matter for function
Identifying druggable binding sites
Designing constructs that actually work in the lab
AlphaFold opened the door. Now we have the tools to walk through it.
From Prediction to Production: How Orbion Completes the Picture
AlphaFold gives you coordinates. Orbion gives you context.
When you upload a protein sequence or AlphaFold structure to Orbion, you get:
1. Complete PTM Landscape
N- and O-glycosylation sites (beyond consensus motifs)
Phosphorylation sites with confidence scores
Lipidation (palmitoylation, myristoylation, prenylation)
Disulfide bonds from structural and evolutionary context
Predictions even when experimental data (UniProt) is absent
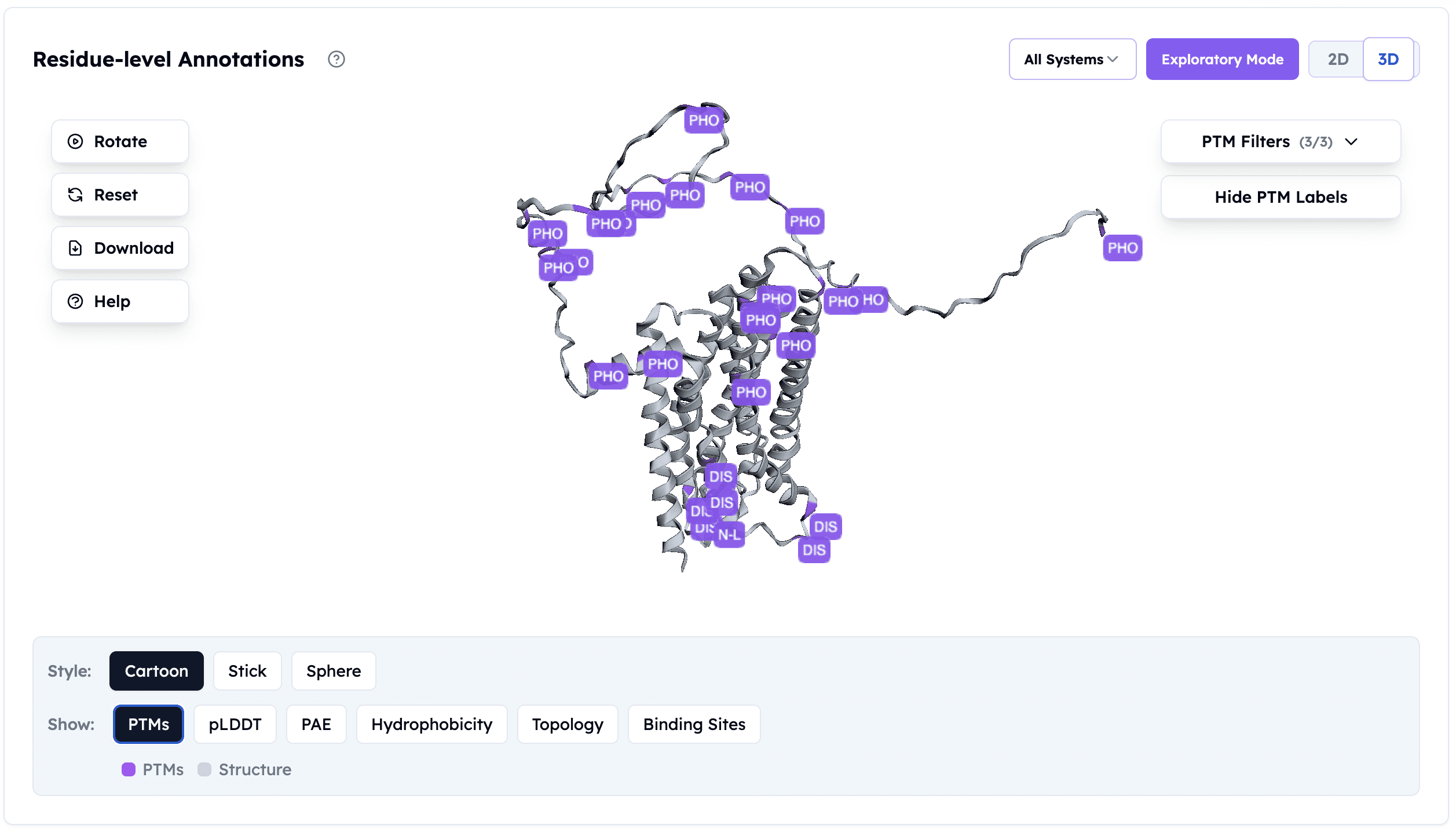
PTM Prediction for Adenosine A2a Receptor: Orbion identifies phosphorylation sites (PHO) and disulfide bonds (DIS) across the GPCR structure — critical information for choosing the right expression system and understanding functional regulation.
2. Binding Site Identification
Active site residues
Allosteric pockets
Protein-protein interfaces
Druggable cavities
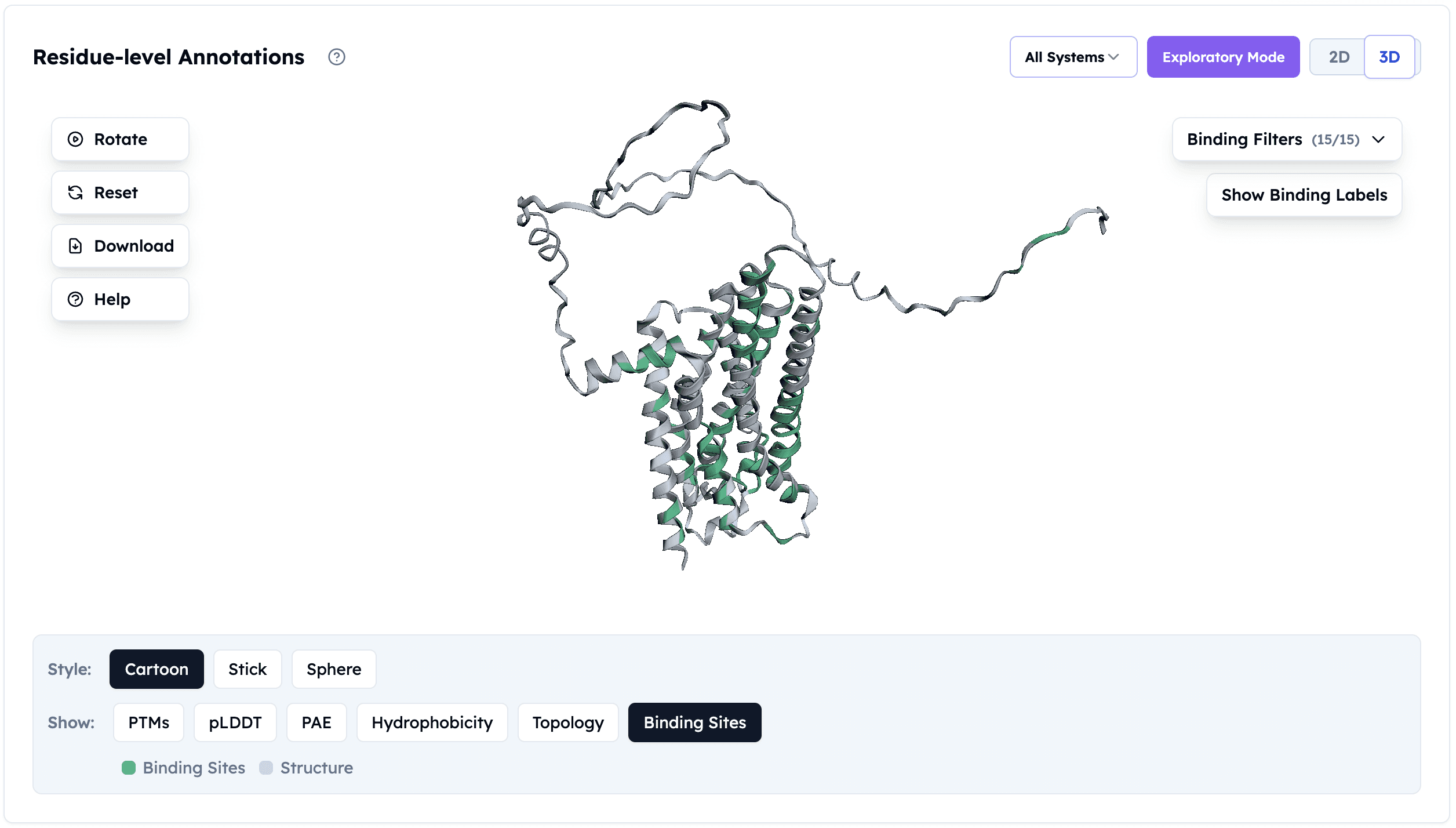
Binding Site Identification: AI-predicted ligand binding residues (green) within the orthosteric pocket of Adenosine A2a receptor. Identifying these interaction interfaces is essential for structure-based drug design and understanding receptor pharmacology.
3. Membrane Topology & Localization
Transmembrane helix boundaries
Intracellular vs extracellular domain annotation
Signal peptide detection
Tag placement recommendations
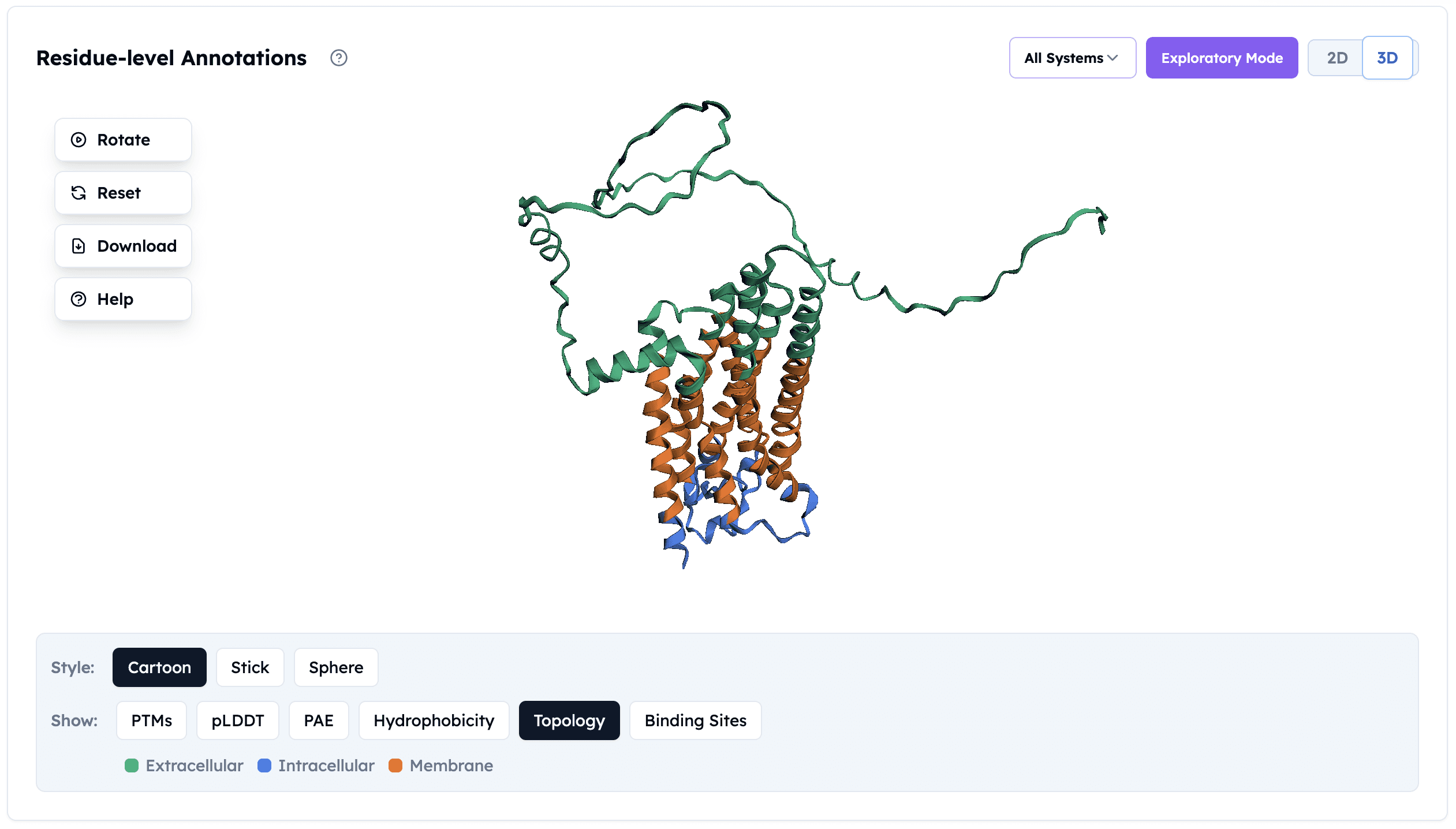
Membrane Topology Annotation: Color-coded visualization showing extracellular loops (green), 7-transmembrane helices (orange), and intracellular regions (blue). Accurate topology prediction guides construct design, tag placement, and purification strategy.
4. Stability Optimization
Thermostabilizing mutation predictions (ΔΔG)
Aggregation hotspot identification
Expression troubleshooting guidance
Impact: Turn an unstable target into a stable, expressible protein.
5. Automated Expression System Recommendations
Based on the complete PTM profile:
Detects glycosylation → rules out E. coli
Identifies complex disulfides → suggests yeast or higher
Recognizes membrane topology → recommends insect or mammalian
Flags therapeutic context → prioritizes CHO/HEK293
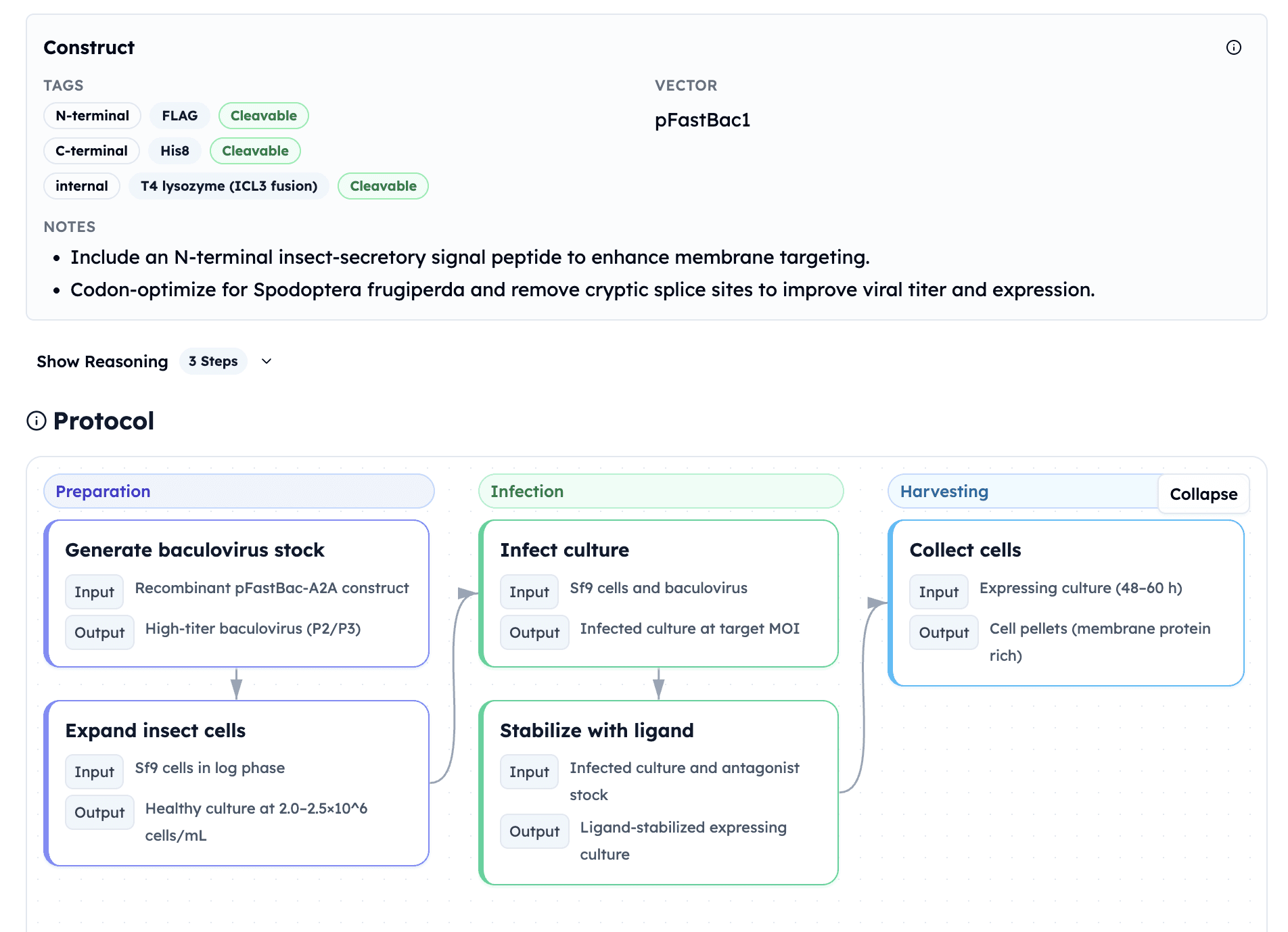
Automated Protocol Generation: Orbion recommends insect cell expression (Sf9/pFastBac1) with T4 lysozyme ICL3 fusion for receptor stabilization—a similar engineering strategy that enabled Brian Kobilka's Nobel Prize-winning Beta-2-AR structure determination.
The Bottom Line
AlphaFold didn't end the race. It moved the starting line.
We no longer spend years wondering what the fold looks like. Now, we have the fold in minutes. The challenge—and the opportunity—lies in what we do next.
By combining the raw power of AlphaFold with tools that explore dynamics (MSA subsampling, Multimer) and biological context (Orbion), we're finally building a complete, high-resolution picture of the druggable proteome.
Structure is solved. Context is next.
Book a 20-Minute Demo
Sign up free for unlimited Overview runs — summary, sequence-based analysis, homology search. For the full Characterization — PTMs, binding sites, stability variants, construct design — book a demo and we'll run your target live.
