Blog
Orbion Team
Detergent-Free Membrane Protein Purification: Nanodiscs and Beyond

You've screened detergents. You found one that solubilizes your GPCR without aggregation. But there's a lingering concern: Is your protein really native-like in a detergent micelle? Are you losing activity? Could the detergent be interfering with your structural studies or functional assays?
Enter detergent-free purification methods: nanodiscs, SMALPs, and amphipols. These approaches keep membrane proteins in a native-like lipid environment (or a stabilizing polymer belt) without the complications of detergent micelles. Here's how they work, when to use them, and how to prepare samples for Cryo-EM.
Key Takeaways
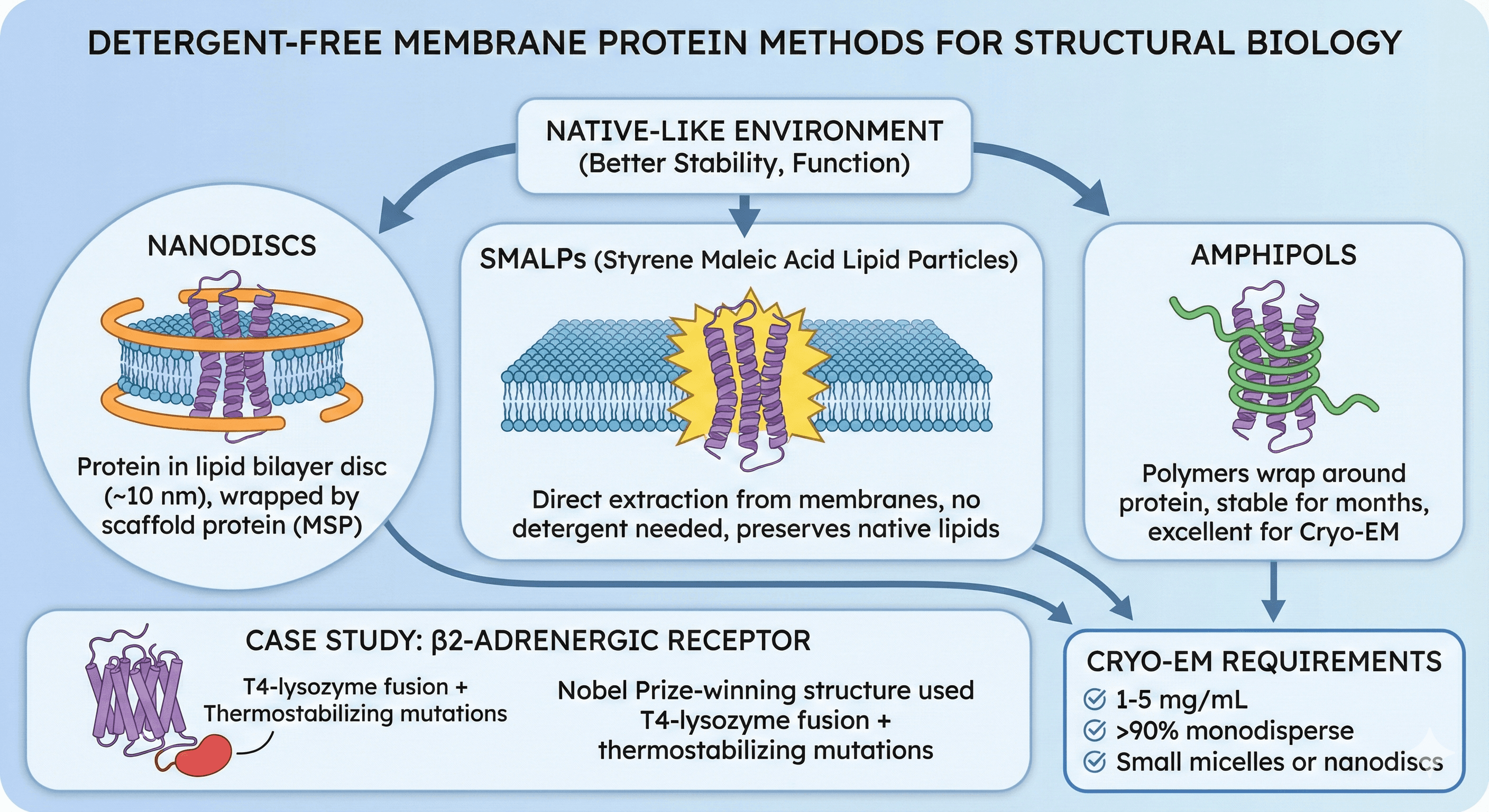
Detergent-free methods maintain native-like environment (better stability, function)
Nanodiscs: Protein in lipid bilayer disc (~10 nm), wrapped by scaffold protein (MSP)
SMALPs: Direct extraction from membranes, no detergent needed, preserves native lipids
Amphipols: Polymers wrap around protein, stable for months, excellent for Cryo-EM
β2-adrenergic receptor: Nobel Prize-winning structure used T4-lysozyme fusion + thermostabilizing mutations
Cryo-EM requires: 1-5 mg/mL, >90% monodisperse, small micelles or nanodiscs
Why Detergent-Free Matters
Problems with Detergents
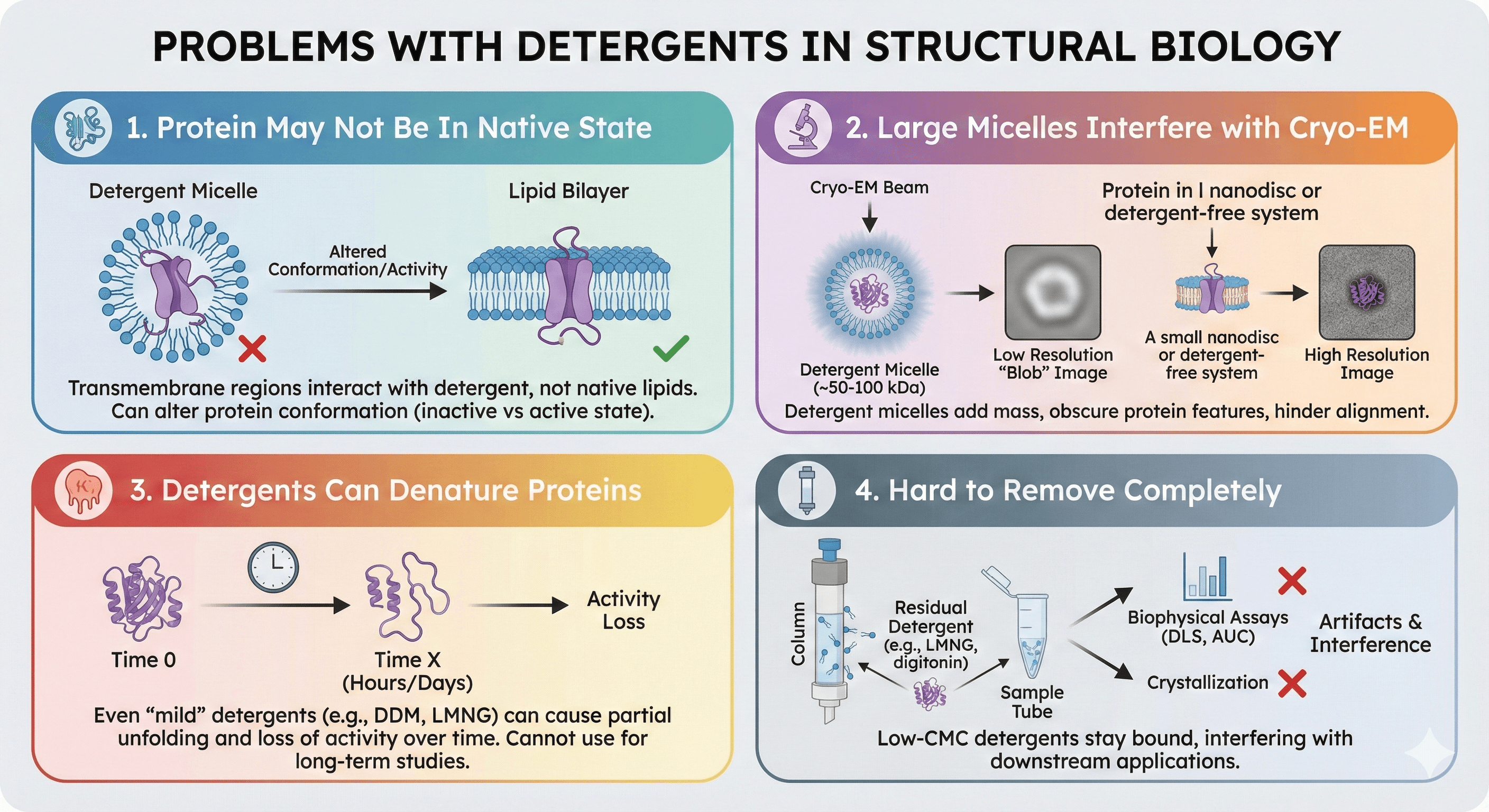
1. Protein may not be in native state
Detergent micelles ≠ lipid bilayer
Transmembrane regions interact with detergent, not native lipids
Can alter protein conformation (inactive state vs active state)
2. Large micelles interfere with Cryo-EM
Detergent micelles add ~50-100 kDa mass around protein
Obscures protein features (lower resolution)
Particle alignment difficult (micelle creates "blob")
3. Detergents can denature proteins
Even "mild" detergents (DDM, LMNG) cause partial unfolding for some proteins
Activity loss over time (hours to days)
Cannot use for long-term studies
4. Hard to remove completely
Low-CMC detergents (LMNG, digitonin) stay bound
Interferes with crystallization, biophysical assays
Detergent in sample causes artifacts (DLS, AUC)
Detergent-Free Approaches Solve These Problems
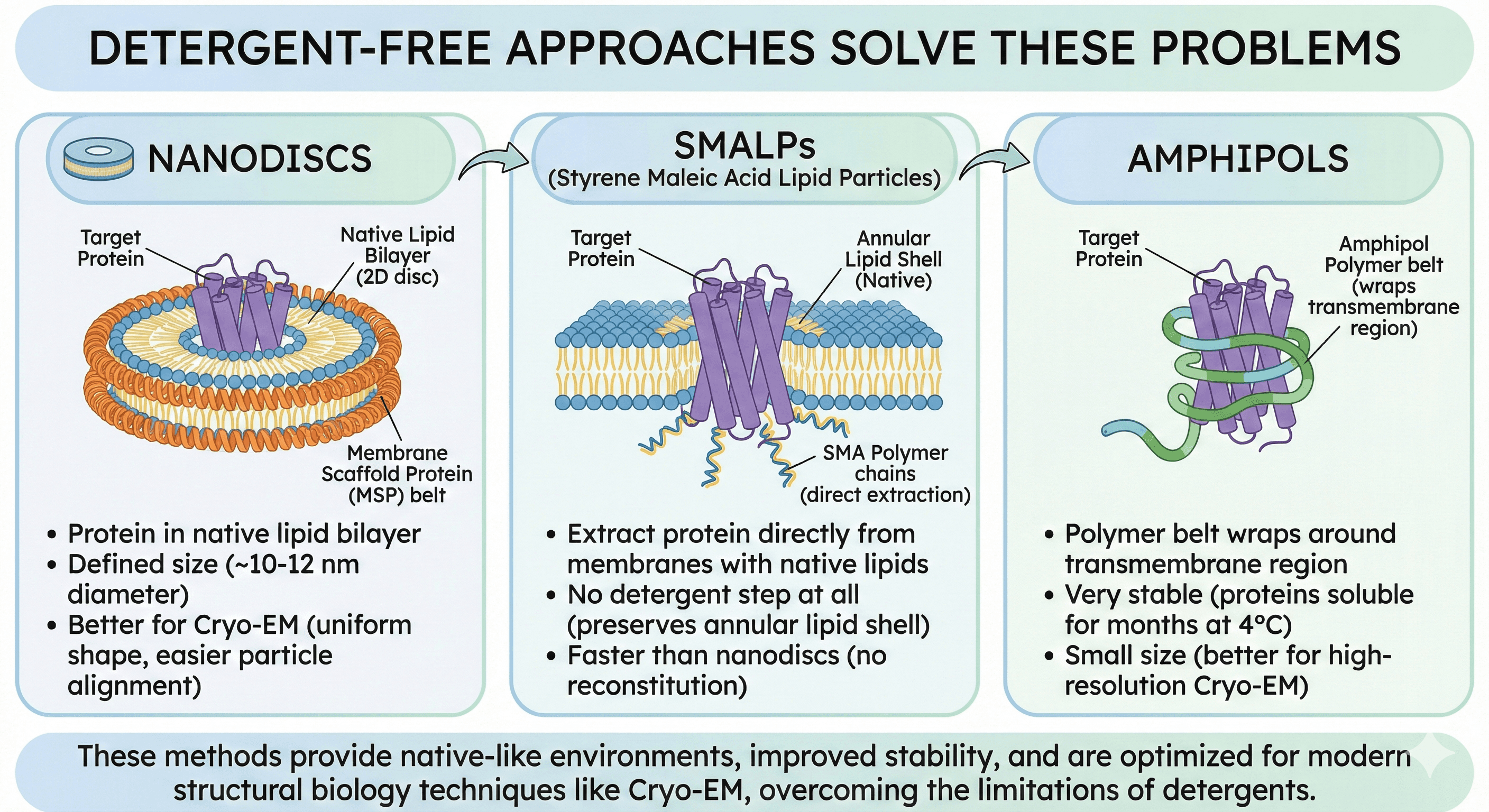
Nanodiscs:
Protein in native lipid bilayer (2D disc)
Defined size (~10-12 nm diameter)
Better for Cryo-EM (uniform shape, easier particle alignment)
SMALPs:
Extract protein directly from membranes with native lipids
No detergent step at all (preserves annular lipid shell)
Faster than nanodiscs (no reconstitution)
Amphipols:
Polymer belt wraps around transmembrane region
Very stable (proteins soluble for months at 4°C)
Small size (better for high-resolution Cryo-EM)
Method 1: Nanodiscs (Lipid Bilayer in a Disc)
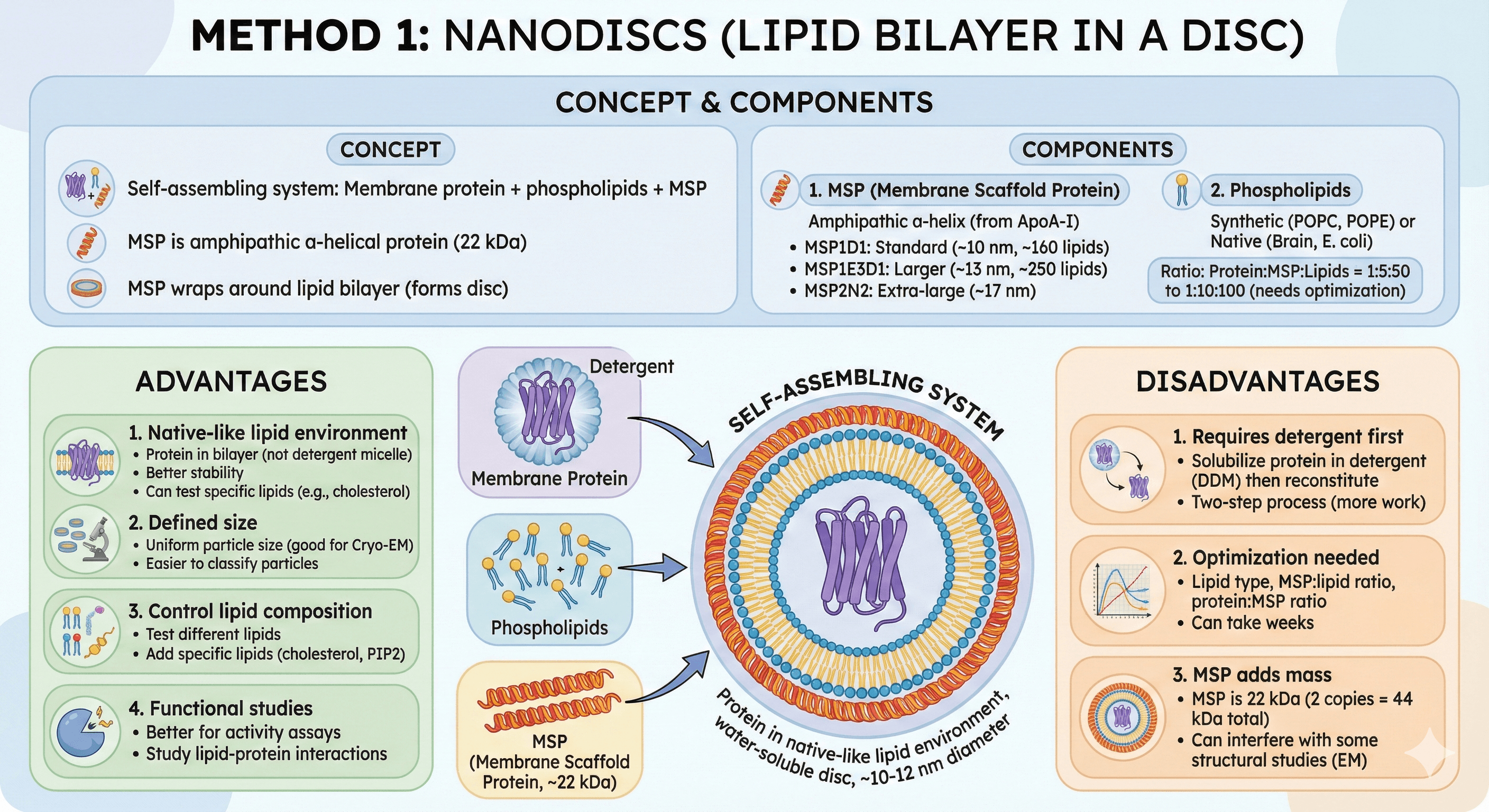
Concept
Self-assembling system:
Membrane protein + phospholipids + MSP (membrane scaffold protein)
MSP is an amphipathic α-helical protein (22 kDa)
MSP wraps around lipid bilayer (forms disc, ~10-12 nm diameter)
Protein sits in native-like lipid environment, but disc is water-soluble
Components
1. MSP (Membrane Scaffold Protein)
Amphipathic α-helix (derived from apolipoprotein A-I)
Wraps around edge of lipid bilayer
Sizes:
MSP1D1: Standard (~10 nm diameter, holds ~160 lipids)
MSP1E3D1: Larger (~13 nm diameter, holds ~250 lipids, for bigger proteins)
MSP2N2: Extra-large (~17 nm, for large complexes)
2. Phospholipids
Synthetic lipids: POPC, POPE, POPG, POPS
Native lipids: Brain polar lipids, E. coli polar lipids
Ratio: Protein:MSP:Lipids = 1:5:50 to 1:10:100 (needs optimization)
Advantages
1. Native-like lipid environment
Protein in bilayer (not detergent micelle)
Better stability, closer to physiological state
Can test specific lipid requirements (e.g., cholesterol for GPCRs)
2. Defined size
Uniform particle size (good for Cryo-EM)
Easier to classify particles (clear shape in EM images)
3. Control lipid composition
Test different lipids systematically
Add specific lipids (cholesterol, cardiolipin, PIP2)
4. Functional studies
Better for activity assays (native-like environment)
Can study lipid-protein interactions
Disadvantages
1. Requires detergent first
Solubilize protein in detergent (DDM)
Then reconstitute into nanodiscs
Two-step process (more work than SMALPs)
2. Optimization needed
Lipid type, MSP:lipid ratio, protein:MSP ratio
Can take weeks to optimize
3. MSP adds mass
MSP is 22 kDa (2 copies per disc = 44 kDa total)
Can interfere with some structural studies
Need to account for MSP mass in EM
Protocol: Nanodisc Reconstitution
Starting material: Protein purified in detergent (DDM, from Part 1)
Step 1: Prepare Components
Protein:
Concentrate to 5-10 mg/mL (in DDM-containing buffer)
Keep at 4°C
MSP:
Express MSP1D1 in E. coli (plasmid available from Addgene)
Purify by His-tag (MSP has N-terminal His-tag)
Store at -80°C in 20 mM Tris pH 7.5, 100 mM NaCl
Lipids:
POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) from Avanti Polar Lipids
Dissolve in chloroform (25 mg/mL stock)
Store at -20°C under argon
Step 2: Mix Components
Typical reaction (100 μL):
Protein: 1 μM (1 nmol)
MSP1D1: 10 μM (10 nmol, 10× excess)
POPC: 100 μM (100 nmol, 100× excess, ~100 lipids per nanodisc)
DDM: 0.5% (in buffer)
Buffer:
20 mM Tris pH 7.5, 150 mM NaCl, 10% glycerol
Calculation notes:
Ratio optimization: Test ratios of protein:MSP:lipids = 1:5:50, 1:10:100, 1:15:150
MSP excess ensures all protein is incorporated
Step 3: Remove Detergent (Critical Step)
Method: Bio-Beads SM-2 (polystyrene resin)
Add 0.5-1 g Bio-Beads per 1 mL reaction (wet weight)
Incubate overnight (12-16 hours) at 4°C with gentle rotation
Bio-Beads absorb DDM → nanodisc self-assembles as detergent is removed
Alternative: Dialysis
Dialyze against 1000× volume of detergent-free buffer
Change buffer 3-4 times (every 4-6 hours)
Slower than Bio-Beads, but works
Step 4: Remove Bio-Beads
Transfer reaction to new tube (leave Bio-Beads behind)
Or centrifuge briefly (1,000 × g, 5 min) and collect supernatant
Step 5: Purify Nanodiscs by SEC
Why: Separate protein-containing nanodiscs from empty nanodiscs
Column: Superdex 200 Increase 10/300 GL
Buffer: 20 mM Tris pH 7.5, 150 mM NaCl, 10% glycerol (no detergent)
Expected elution:
Protein-containing nanodiscs: Elute earlier (~12-14 mL, larger MW)
Empty nanodiscs: Elute later (~15-16 mL, smaller MW)
Collect: Fractions from protein-containing peak
Step 6: Characterization
TEM (Transmission Electron Microscopy):
Negative stain with uranyl acetate
Expected: Circular discs visible (~10-12 nm diameter)
Confirms nanodisc formation
SEC-MALS:
Measure MW of nanodisc
Expected: Protein MW + 2× MSP (44 kDa) + lipids (~120 kDa total for 50 kDa protein)
Activity assay:
Test ligand binding or transport activity
Compare to detergent-solubilized protein
Troubleshooting Nanodiscs
Problem: No protein in nanodiscs (empty nanodiscs only)
Cause: Wrong lipid:MSP ratio, or protein aggregated during reconstitution
Solution: Optimize ratio, add more protein, keep cold (4°C)
Problem: Heterogeneous sample (multiple peaks in SEC)
Cause: Multiple oligomeric states, or variable nanodisc sizes
Solution: Optimize MSP:lipid ratio, use size-defined MSP (MSP1E3D1 for larger proteins)
Problem: Protein inactive in nanodiscs
Cause: Wrong lipid type, or lipid:protein ratio too low
Solution: Test different lipids (add cholesterol for GPCRs, cardiolipin for respiratory complexes)
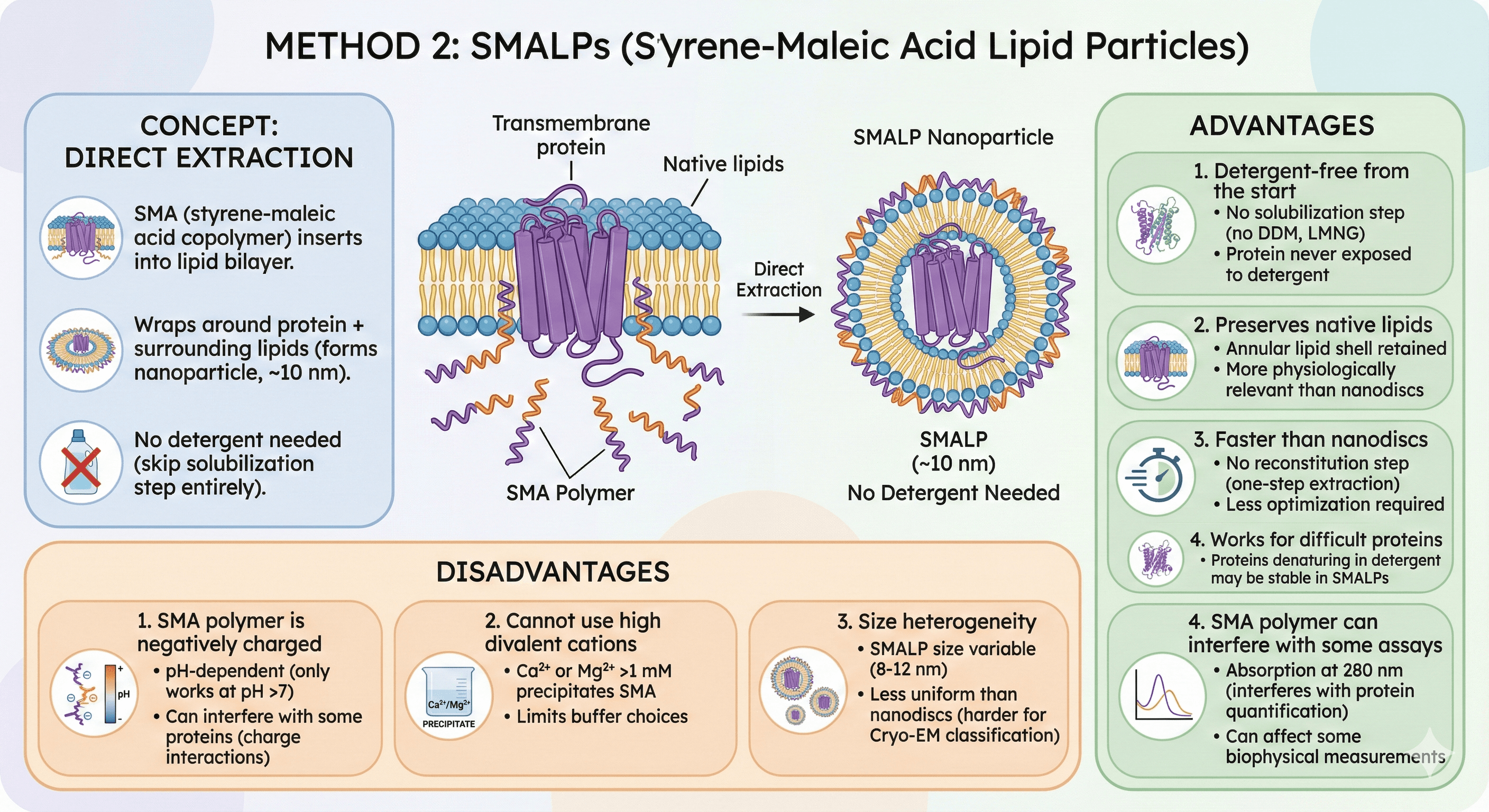
Method 2: SMALPs (Styrene-Maleic Acid Lipid Particles)
Concept
Direct extraction from membranes:
SMA (styrene-maleic acid copolymer) inserts into lipid bilayer
Wraps around protein + surrounding lipids (forms nanoparticle, ~10 nm)
No detergent needed (skip solubilization step entirely)
Advantages
1. Detergent-free from the start
No solubilization step (no DDM, no LMNG)
Protein never exposed to detergent
2. Preserves native lipids
Annular lipid shell around protein retained
More physiologically relevant than nanodiscs with synthetic lipids
3. Faster than nanodiscs
No reconstitution step (one-step extraction)
Less optimization required
4. Works for difficult proteins
Proteins that denature in detergent may be stable in SMALPs
Disadvantages
1. SMA polymer is negatively charged
pH-dependent (only works at pH >7)
Can interfere with some proteins (charge interactions)
2. Cannot use high divalent cations
Ca²⁺ or Mg²⁺ >1 mM precipitates SMA
Limits buffer choices
3. Size heterogeneity
SMALP size variable (8-12 nm)
Less uniform than nanodiscs (harder for Cryo-EM classification)
4. SMA polymer can interfere with some assays
Absorption at 280 nm (interferes with protein quantification)
Can affect some biophysical measurements
Protocol: SMALP Extraction
Starting material: Crude membranes (from Part 1, Stage 2)
Step 1: Add SMA Polymer to Membranes
Membranes:
Resuspend to 10 mg/mL total protein (in buffer)
SMA polymer:
Add to 2.5% (w/v) final concentration
Ratio: 1:2.5 (protein:SMA, typical starting point)
Buffer:
50 mM Tris pH 7.5 or 50 mM HEPES pH 7.5 (pH must be >7 for SMA to work)
150 mM NaCl
Protease inhibitors
No Ca²⁺ or Mg²⁺ >1 mM (precipitates SMA)
Incubate:
2 hours at room temperature (RT) or 4 hours at 4°C
Gentle mixing (rotator or nutator)
Step 2: Clarify
Centrifugation:
100,000 × g, 30 min, 4°C
Supernatant: Protein in SMALPs (solubilized) Pellet: Unsolubilized membrane (discard)
Step 3: Affinity Purification
Same as detergent protocol (Part 1), but no detergent in buffers:
Wash buffer:
50 mM Tris pH 7.5, 150 mM NaCl
20 mM imidazole
10% glycerol
No detergent
Elution buffer:
50 mM Tris pH 7.5, 150 mM NaCl
250 mM imidazole
10% glycerol
Step 4: SEC (Polishing)
Column: Superdex 200
Buffer: 20 mM Tris pH 7.5, 150 mM NaCl, 10% glycerol (no detergent)
Expected: Single peak (protein in SMALP)
Step 5: Characterization
TEM: Observe SMALP particles (circular, ~10 nm)
Activity assay: Test function (compare to detergent)
SEC-MALS: Measure MW (protein + SMA + lipids)
Troubleshooting SMALPs
Problem: Low solubilization efficiency (<20%)
Cause: SMA:protein ratio too low, or pH too low
Solution: Increase SMA (2.5% → 5%), check pH >7.5
Problem: Precipitate forms during incubation
Cause: Ca²⁺ or Mg²⁺ in buffer
Solution: Use Ca²⁺/Mg²⁺-free buffer, add EGTA (1 mM) to chelate residual Ca²⁺
Problem: Protein inactive in SMALPs
Cause: SMA polymer interferes with functional site
Solution: Try alternative polymers (DIBMA, SMA-QA with different charge properties)
Method 3: Amphipols (Amphipathic Polymers)
Concept
Polymer belt wraps around transmembrane region:
Amphipols are amphipathic polymers (hydrophobic + hydrophilic segments)
Wrap around protein like a belt (shield TM helices)
Protein soluble in aqueous buffer, no detergent needed
Types of Amphipols
A8-35 (most common):
Poly(acrylic acid) backbone with octyl side chains
MW ~4-5 kDa per polymer chain
Negatively charged (works best at pH 7-8)
PMAL-C8:
Poly(maleic acid) with octyl chains
Alternative to A8-35
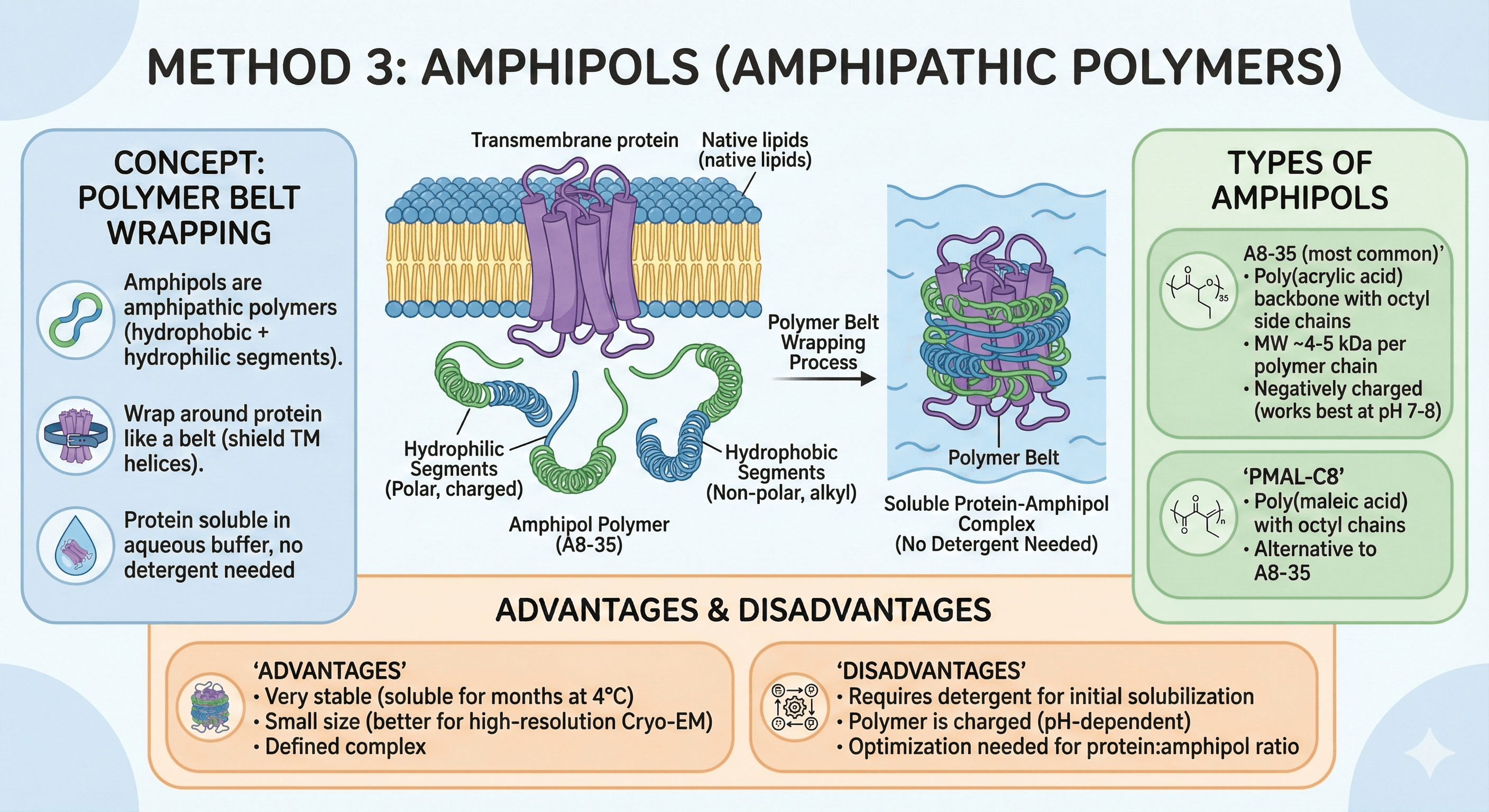
Advantages
1. No CMC (don't form micelles)
Stay bound to protein (don't dialyze out)
Stable association
2. Very stable
Proteins remain soluble for months at 4°C
Better than detergents (no slow aggregation)
3. Small size
Amphipol belt is thin (~3-4 nm)
Better for high-resolution Cryo-EM (less obscuring)
4. Compatible with many biophysical techniques
NMR (smaller than detergent micelles)
Cryo-EM (excellent, minimal background)
Disadvantages
1. Expensive
~$100-200/g (more than most detergents)
2. Can be hard to remove
If you need to later (e.g., for lipid reconstitution), amphipols are difficult to remove
3. Can block functional sites
Polymer may sterically block binding sites or conformational changes
Not ideal for all functional assays
Protocol: Amphipol Exchange
Starting material: Protein in detergent (DDM or LMNG)
Step 1: Mix Protein with Amphipol
Ratio: 1:5 (protein:amphipol, weight ratio)
Example: 1 mg protein + 5 mg amphipol A8-35
Incubate: 2 hours at 4°C (gentle mixing)
Step 2: Remove Detergent
Method 1: Bio-Beads
Add Bio-Beads (0.5 g per mL)
Incubate overnight at 4°C
Bio-Beads absorb detergent → amphipol remains bound to protein
Method 2: Dialysis
Dialyze against amphipol-free buffer (1000× volume)
Change buffer 3-4 times
Step 3: Remove Free Amphipol (Optional)
SEC: Superdex 200
Protein-amphipol complex elutes earlier (higher MW)
Free amphipol elutes later
Step 4: Characterization
DLS: Measure particle size (Rh should be small, ~5-8 nm for 50 kDa protein)
SEC-MALS: Confirm MW (protein + amphipol)
Activity assay: Test if protein is functional in amphipols
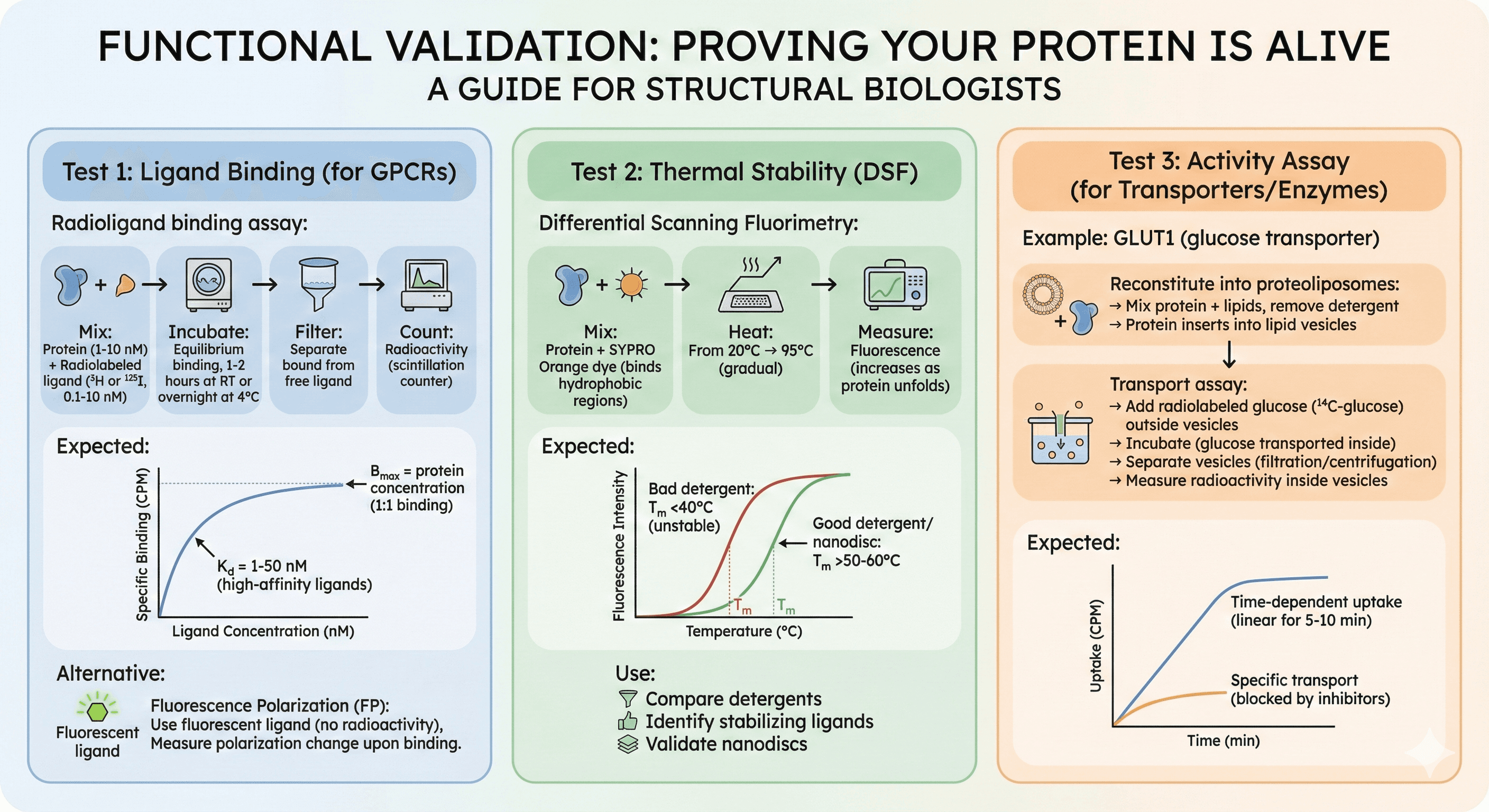
Functional Validation: Proving Your Protein Is Alive
Test 1: Ligand Binding (for GPCRs)
Radioligand binding assay:
Mix protein (1-10 nM) + radiolabeled ligand (³H or ¹²⁵I, 0.1-10 nM)
Incubate (equilibrium binding, 1-2 hours at RT or overnight at 4°C)
Filter (separate bound from free ligand)
Count radioactivity (scintillation counter)
Expected:
Specific binding: Kd = 1-50 nM (high-affinity ligands)
Bmax = protein concentration (1:1 binding)
Alternative: Fluorescence Polarization (FP)
Use fluorescent ligand (no radioactivity)
Measure polarization change upon binding
Test 2: Thermal Stability (DSF)
Differential Scanning Fluorimetry:
Mix protein + SYPRO Orange dye (binds hydrophobic regions)
Heat from 20°C → 95°C (gradual)
Measure fluorescence (increases as protein unfolds)
Expected:
Tm (melting temperature): Inflection point
Good detergent/nanodisc: Tm >50-60°C
Bad detergent: Tm <40°C (unstable)
Use: Compare detergents, identify stabilizing ligands, validate nanodiscs
Test 3: Activity Assay (for Transporters/Enzymes)
Example: GLUT1 (glucose transporter)
Reconstitute into proteoliposomes:
Mix protein + lipids, remove detergent
Protein inserts into lipid vesicles (liposomes)
Transport assay:
Add radiolabeled glucose (¹⁴C-glucose) outside vesicles
Incubate (glucose transported inside)
Separate vesicles (filtration or centrifugation)
Measure radioactivity inside vesicles
Expected:
Time-dependent uptake (linear for 5-10 min)
Specific transport (blocked by inhibitors)
Case Study: β2-Adrenergic Receptor (Nobel Prize-Winning Purification)
The Challenge
Target: β2-adrenergic receptor (GPCR, 7 TM helices)
Historical context:
GPCRs extremely difficult to crystallize (unstable, flexible)
No GPCR structure until 2007 (despite decades of effort)
The Solution (Brian Kobilka's Lab)
Expression: Sf9 insect cells, baculovirus (5-10 mg/L yield)
Construct Engineering (Critical for Success):
1. N-terminal truncation
Removed disordered region (residues 1-33)
Reduces flexibility
2. ICL3 replacement
Replaced flexible intracellular loop 3 (ICL3) with T4 lysozyme
T4-lysozyme acts as fiducial marker for crystallization (provides crystal contacts)
Rigidifies receptor
3. C-terminal truncation
Removed phosphorylation sites (residues 365-413)
Reduces heterogeneity
4. Thermostabilizing mutations
6 point mutations (identified by alanine scanning)
Increased Tm by ~20°C (from 40°C → 60°C)
Mutations: L48A, E122W, Y132G, R149L, Y219A, H296A
Purification:
1. Membrane preparation: Ultracentrifugation
2. Solubilization:
1% DDM + 0.2% CHS (cholesteryl hemisuccinate, stabilizing lipid)
3. Affinity:
Ni-NTA (His-tag at C-terminus)
4. Ligand binding:
Add alprenolol (inverse agonist, 10 μM)
Stabilizes inactive state (locks conformation)
5. SEC:
Superdex 200
Monodisperse peak (single, sharp)
6. Concentrate:
To 40-50 mg/mL (for crystallization)
Crystallization:
Lipidic cubic phase (LCP): Protein in monoolein lipid matrix
Crystals grown over weeks
X-ray diffraction: 2.4 Å resolution
Impact:
First high-resolution GPCR structure (2007, Nature)
Brian Kobilka awarded Nobel Prize (2012, with Robert Lefkowitz)
Enabled structure-based drug design for GPCRs (>500 drugs target GPCRs)
Key Lessons:
1. Construct engineering is critical
Truncations, fusions, thermostabilizing mutations = success
2. Ligand stabilization matters
Inverse agonist (alprenolol) locks receptor in stable conformation
Without ligand: Receptor is too flexible, cannot crystallize
3. Lipid matters
CHS (cholesteryl hemisuccinate) stabilizes GPCR
Mimics cholesterol (native lipid for GPCRs)
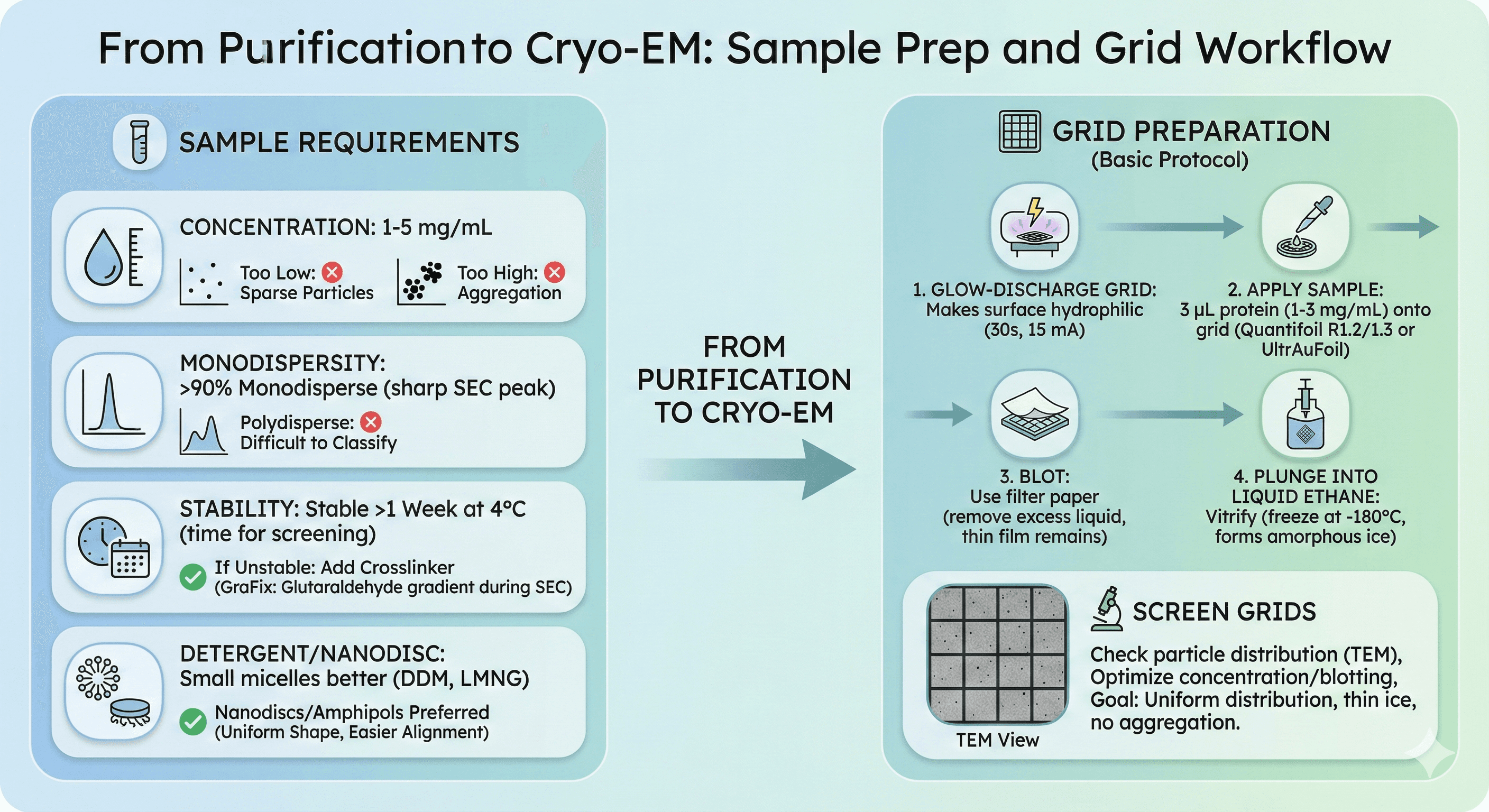
From Purification to Cryo-EM
Sample Requirements
Concentration:
1-5 mg/mL (optimal for grid preparation)
Too low: Particles sparse
Too high: Aggregation on air-water interface
Monodispersity:
>90% monodisperse (sharp SEC peak)
Polydisperse samples: Difficult to classify particles
Stability:
Stable >1 week at 4°C (time needed for grid screening)
If unstable: Add crosslinker (GraFix: glutaraldehyde gradient during SEC)
Detergent/nanodisc considerations:
Small micelles better: DDM, LMNG (vs Triton X-100, large)
Nanodiscs/amphipols preferred: Uniform shape, easier particle alignment
Grid Preparation (Basic Protocol)
1. Glow-discharge grid
Makes surface hydrophilic (30 seconds, 15 mA)
2. Apply sample
3 μL protein (1-3 mg/mL)
Onto grid (Quantifoil R1.2/1.3 or UltrAuFoil)
3. Blot
Use filter paper (remove excess liquid, thin film remains)
4. Plunge into liquid ethane
Vitrify (freeze at -180°C, forms amorphous ice)
Screen grids:
Check particle distribution (TEM)
Optimize concentration, blotting time
Goal: Uniform particle distribution, thin ice, no aggregation
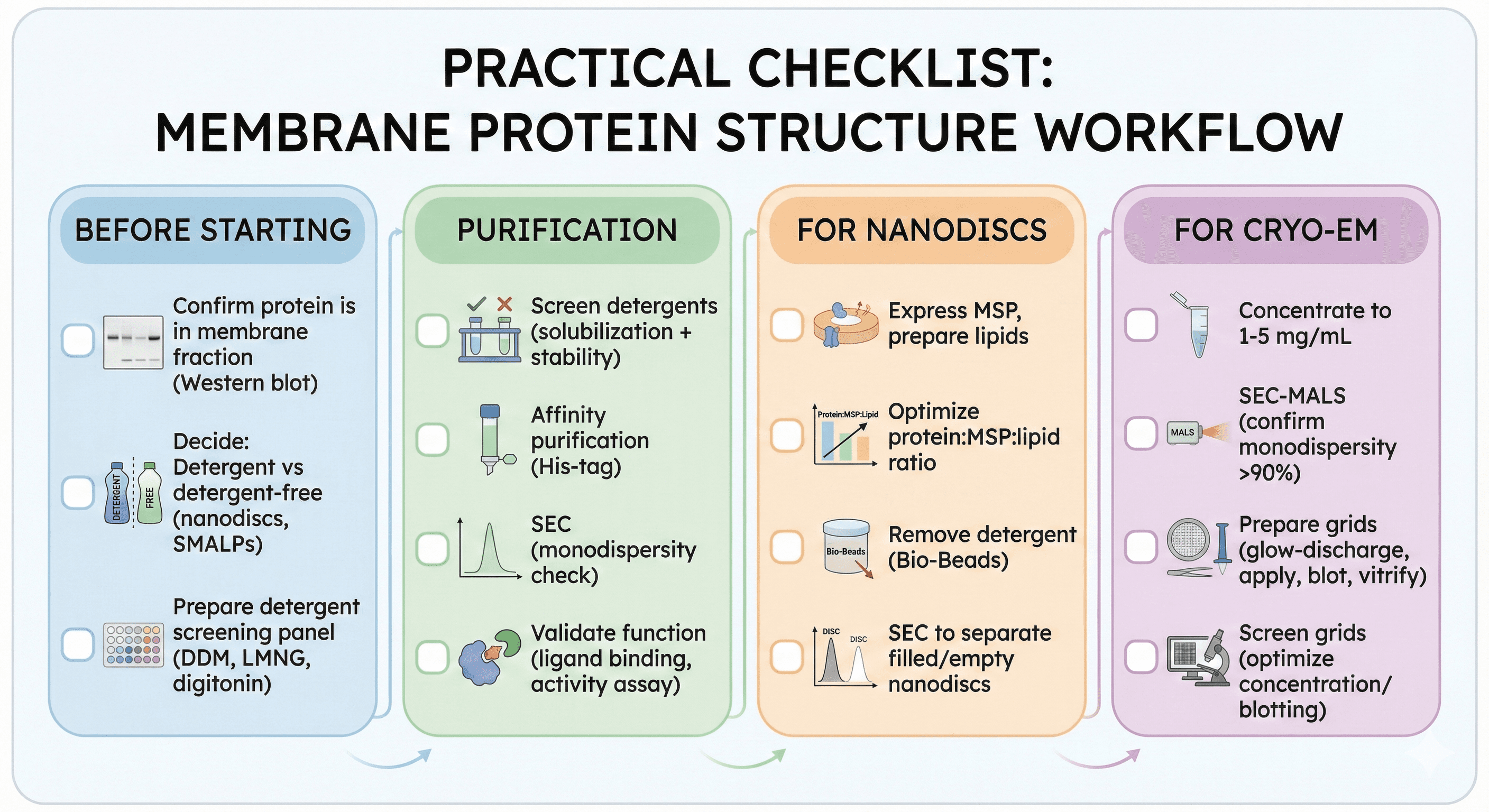
Practical Checklist
Before Starting
[ ] Confirm protein is in membrane fraction (Western blot)
[ ] Decide: Detergent vs detergent-free (nanodiscs, SMALPs)
[ ] Prepare detergent screening panel (DDM, LMNG, digitonin)
Purification
[ ] Screen detergents (solubilization + stability)
[ ] Affinity purification (His-tag)
[ ] SEC (monodispersity check)
[ ] Validate function (ligand binding, activity assay)
For Nanodiscs
[ ] Express MSP, prepare lipids
[ ] Optimize protein:MSP:lipid ratio
[ ] Remove detergent (Bio-Beads)
[ ] SEC to separate filled/empty nanodiscs
For Cryo-EM
[ ] Concentrate to 1-5 mg/mL
[ ] SEC-MALS (confirm monodispersity >90%)
[ ] Prepare grids (glow-discharge, apply, blot, vitrify)
[ ] Screen grids (optimize concentration/blotting)
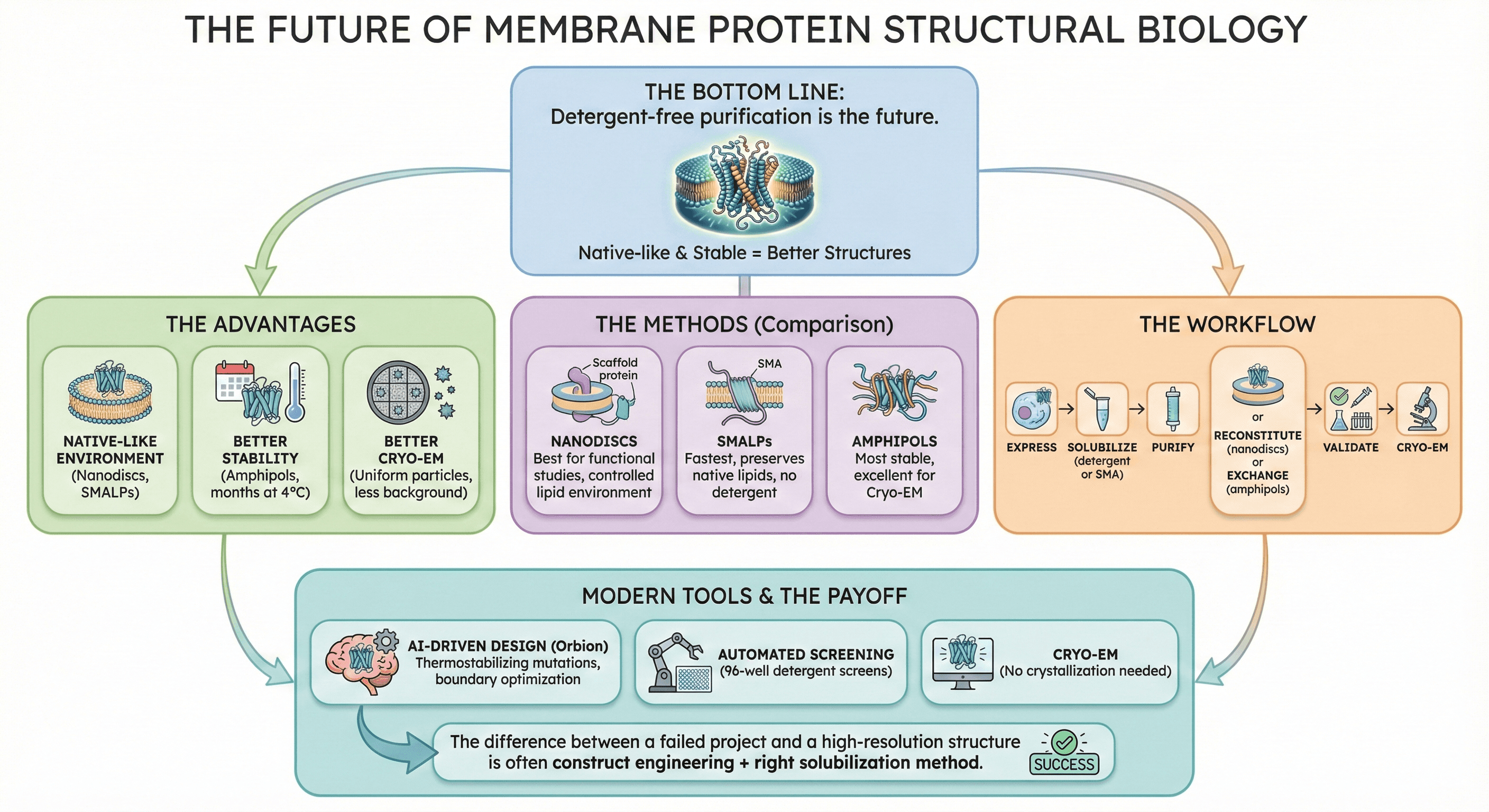
The Bottom Line
Detergent-free purification is the future of membrane protein structural biology.
The advantages:
Native-like environment (nanodiscs, SMALPs)
Better stability (amphipols, months at 4°C)
Better Cryo-EM (uniform particles, less background)
The methods:
Nanodiscs: Best for functional studies, controlled lipid environment
SMALPs: Fastest, preserves native lipids, no detergent
Amphipols: Most stable, excellent for Cryo-EM
The workflow:
Express → Solubilize (detergent or SMA) → Purify → Reconstitute (nanodiscs) or Exchange (amphipols) → Validate → Cryo-EM
Modern tools accelerate success:
AI-driven construct design (Orbion: thermostabilizing mutations, boundary optimization)
Automated screening (96-well detergent screens)
Cryo-EM (no crystallization needed)
The difference between a failed membrane protein project and a high-resolution structure is often construct engineering + right solubilization method.
Ready to Optimize Your Membrane Protein?
If you're struggling with membrane protein stability, expression, or construct design, Orbion can help.
Orbion provides:
Thermostabilizing mutation predictions (increase Tm, reduce aggregation)
Construct boundary design (truncate disordered regions, identify stable domains)
Membrane topology prediction (TM helices, loop regions, signal peptides)
Expression system recommendations (E. coli vs insect vs mammalian)
PTM prediction (glycosylation sites requiring specific expression systems)
Book a 20-Minute Demo
Sign up free for unlimited Overview runs — summary, sequence-based analysis, homology search. For the full Characterization — PTMs, binding sites, stability variants, construct design — book a demo and we'll run your target live.
