Blog
Orbion Team
Why Your Protein Won't Crystallize: Understanding the 6 Common Failure Modes

You've spent 6 months expressing and purifying your protein. The SEC-MALS looks perfect—monodisperse, no aggregates. Thermal stability is excellent. You set up 96-well crystallization screens. Two weeks later: clear drops. No crystals.
You try another screen. Then another. Different concentrations. Different buffers. Six months pass. Still nothing.
Welcome to the most frustrating bottleneck in structural biology: crystallization. It kills more structural projects than any other technical barrier, and the failure mechanisms are often invisible until you know exactly what to look for.
This is Part 1 of our crystallization troubleshooting guide. Here, we'll diagnose why proteins fail to crystallize. In Part 2, we'll cover how to fix each problem with modern computational tools.
Key Takeaways
70-80% of structural biology projects fail at the crystallization stage
Main culprits: Conformational flexibility (35%), surface entropy (25%), sample heterogeneity (20%), wrong construct boundaries (15%), aggregation (20%), missing cofactors (5%)
Cost of failure: $80-150K wasted per failed target (6-12 months of effort)
The solution: Understanding failure modes before setting up thousands of crystallization trials
Modern approach: AI-driven prediction identifies problems in minutes, not months
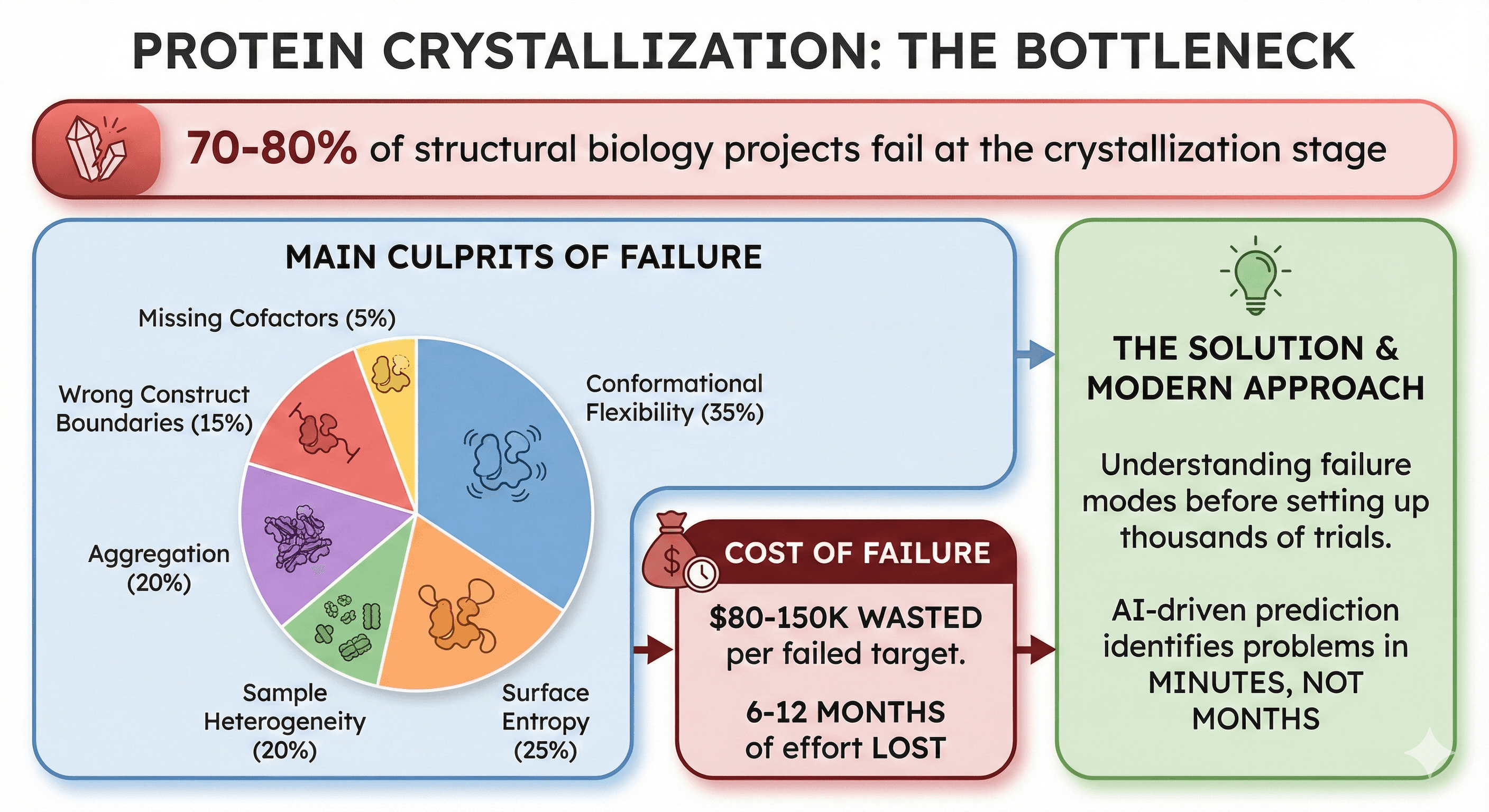
The Crystallization Crisis: By the Numbers
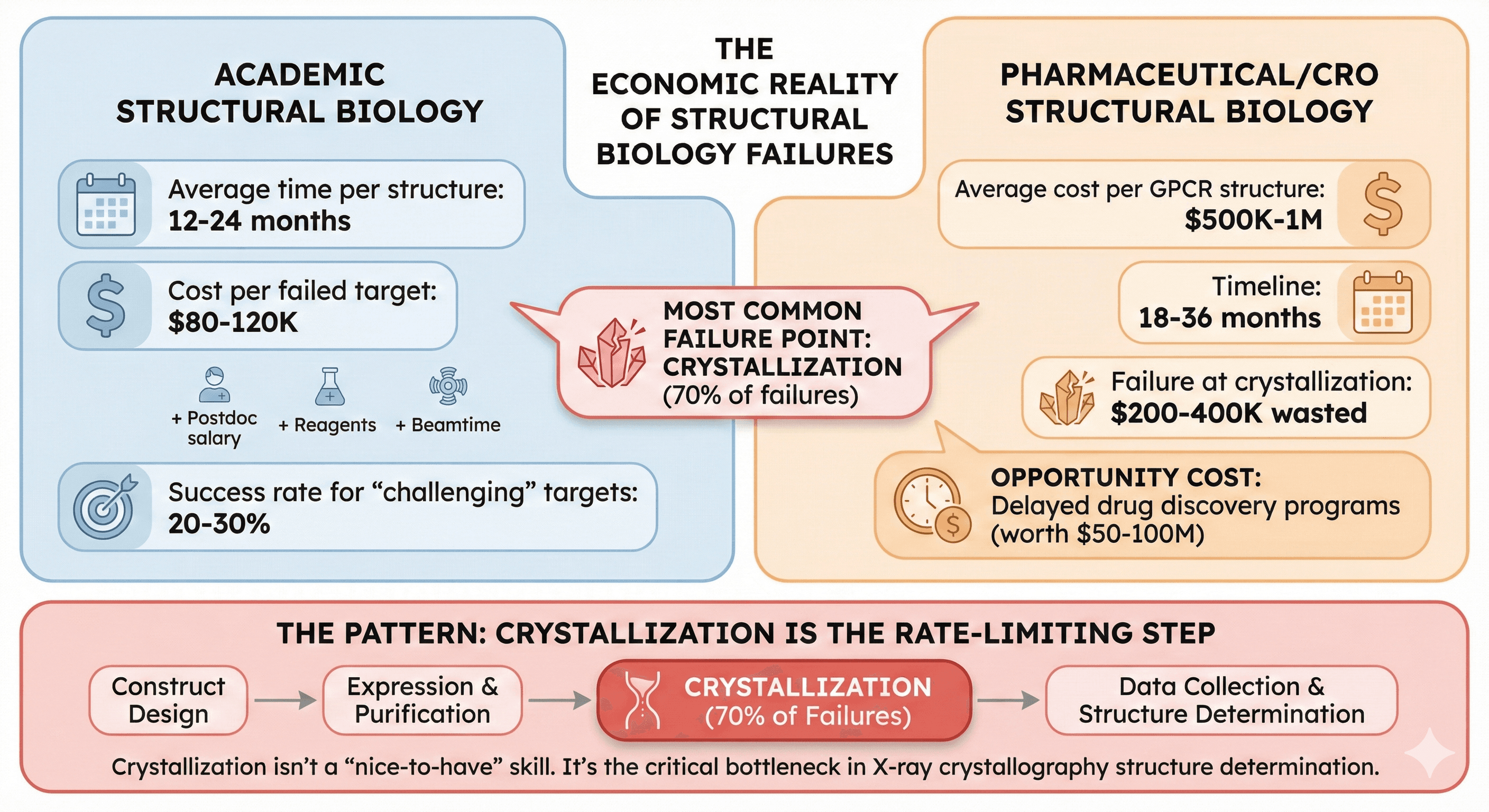
The Economic Reality
Academic structural biology:
Average time per structure: 12-24 months
Cost per failed target: $80-120K (postdoc salary + reagents + beamtime)
Success rate for "challenging" targets: 20-30%
Most common failure point: Crystallization (70% of failures)
Pharmaceutical/CRO structural biology:
Average cost per GPCR structure: $500K-1M
Timeline: 18-36 months
Failure at crystallization: $200-400K wasted
Opportunity cost: Delayed drug discovery programs (worth $50-100M)
The pattern: Crystallization isn't a "nice-to-have" skill. It's the rate-limiting step in structure determination for X-ray crystallography.
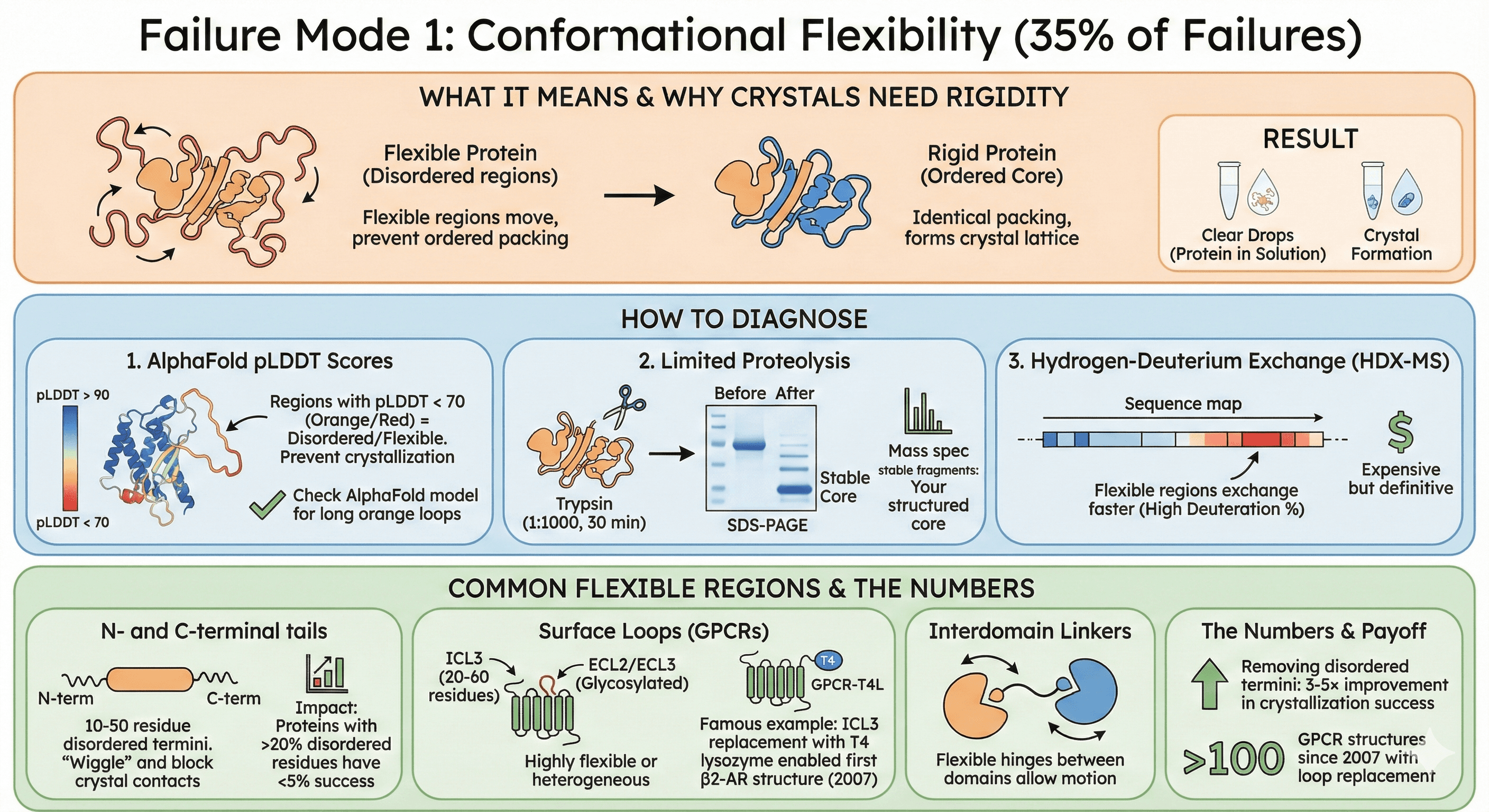
Failure Mode 1: Conformational Flexibility (35% of Failures)
What it means: Your protein has flexible regions (loops, termini, linkers) that move and prevent the tight, ordered packing required for crystal lattice formation.
Why crystals need rigidity:
Crystallization requires identical packing of every protein molecule
Flexible regions adopt different conformations in each molecule
No single, repeatable lattice can form
Result: Protein stays in solution (clear drops)
How to Diagnose
1. AlphaFold pLDDT scores:
Regions with pLDDT < 70 (orange/red) = disordered/flexible
These regions will prevent crystallization
Check your AlphaFold model: Are there long orange loops?
2. Limited proteolysis:
Treat purified protein with low protease (trypsin 1:1000, 30 min)
Run SDS-PAGE: Flexible loops are cleaved first
Mass spec the stable fragments: These are your structured core
3. Hydrogen-deuterium exchange (HDX-MS):
Flexible regions exchange faster (high deuteration %)
Expensive but definitive
Identifies exact residues that are flexible
Common Flexible Regions
N- and C-terminal tails:
Most proteins have 10-50 residue disordered termini
These "wiggle" in solution, blocking crystal contacts
Impact: Proteins with >20% disordered residues have <5% crystallization success
Surface loops (especially in GPCRs):
Intracellular loop 3 (ICL3) in GPCRs: 20-60 residues, highly flexible
Extracellular loops (ECL2, ECL3): Glycosylated, heterogeneous
Famous example: GPCR ICL3 replacement with T4 lysozyme enabled first β2-AR structure (2007)
Interdomain linkers:
Flexible hinges between domains
Allow domain motion (biologically important, structurally problematic)
The numbers:
Removing disordered termini: 3-5× improvement in crystallization success
GPCR loop replacement: Enabled >100 GPCR structures since 2007
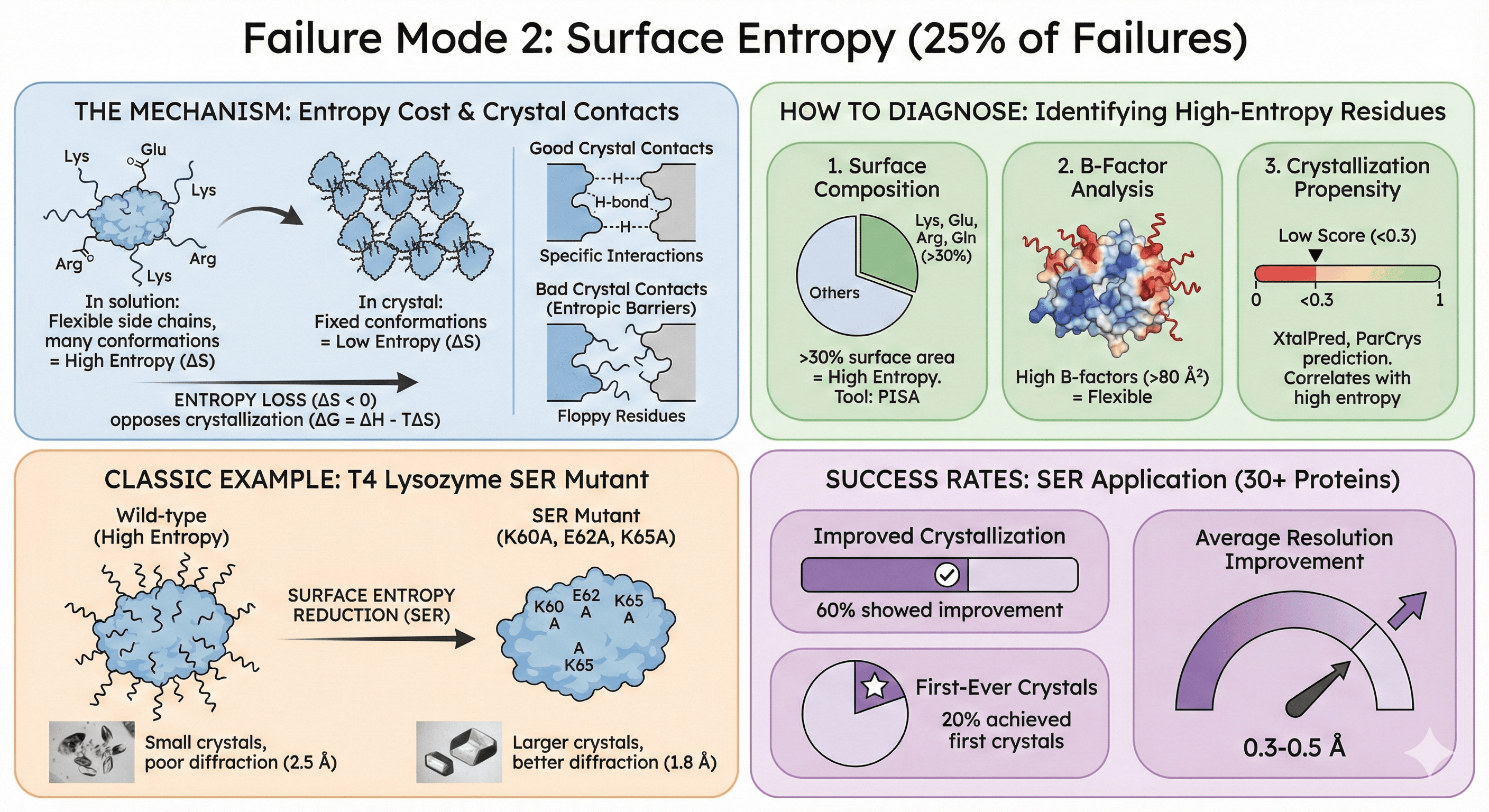
Failure Mode 2: Surface Entropy (25% of Failures)
What it means: High-entropy surface residues (long, flexible side chains like Lys, Glu, Arg) prevent tight crystal packing.
The Mechanism
Entropy cost of crystallization:
In solution: Lys, Glu, Arg side chains are flexible (many conformations = high entropy)
In crystal: These side chains must adopt fixed conformations (low entropy)
Entropy loss opposes crystallization (ΔG = ΔH - TΔS; large negative ΔS makes ΔG unfavorable)
Crystal contacts require specificity:
Good crystal contacts: Complementary surfaces with specific interactions (H-bonds, salt bridges)
Bad crystal contacts: Floppy residues that can't form stable interfaces
High-entropy residues create "entropic barriers" to crystallization
How to Diagnose
1. Surface composition analysis:
Calculate % of surface area occupied by Lys, Glu, Arg, Gln
>30% = high entropy, poor crystallization propensity
Tool: PISA (protein surface analysis)
2. B-factor analysis (if you have homolog structures):
High B-factors (>80 Ų) on surface residues = flexible
These residues will resist crystallization
3. Crystallization propensity prediction:
XtalPred, ParCrys: Predict crystallization likelihood from sequence
Low scores (<0.3) often correlate with high surface entropy
Classic Example: T4 Lysozyme
Wild-type: Difficult to crystallize (small crystals, poor diffraction) SER mutant: K60A, E62A, K65A (three surface mutations) Result: Larger crystals, better diffraction (2.5 Å → 1.8 Å resolution)
Success rates:
SER applied to 30+ proteins: 60% showed improved crystallization
20% achieved first-ever crystals
Average resolution improvement: 0.3-0.5 Å
Failure Mode 3: Sample Heterogeneity (20% of Failures)
What it means: Your "pure" protein is actually a mixture of conformations, oligomeric states, or post-translational modifications. Crystals require homogeneity—every molecule identical.
Source 1: Post-Translational Modifications (PTMs)
The problem:
N-glycosylation: Produces heterogeneous glycan structures (different sizes, branching)
Each glycoform behaves differently in crystallization
Result: 10-20 different species in "pure" sample
Example: GPCR N-glycosylation
Typical GPCR has 2-4 N-glycosylation sites
Each site can have 5-10 different glycan structures
Total glycoforms: 5² to 10⁴ = 25 to 10,000 distinct species
Crystallization: Impossible with this heterogeneity
How to detect:
SDS-PAGE: Glycosylated proteins run as smear (not sharp band)
Mass spectrometry: Multiple peaks separated by ~200-300 Da (sugar units)
Lectin binding: ConA, WGA bind glycans (confirms glycosylation)
Source 2: Conformational Heterogeneity
The problem: Protein exists in multiple conformational states (open/closed, active/inactive, apo/holo).
Example: Kinases
DFG-in (active) vs DFG-out (inactive)
Your sample is a mixture (60% DFG-in, 40% DFG-out)
Neither conformation can form ordered crystals alone
Source 3: Oligomeric State Heterogeneity
The problem: Protein exists as mixture of monomers, dimers, tetramers.
How to detect:
SEC-MALS: Multiple peaks (monomer at 50 kDa, dimer at 100 kDa)
Native PAGE: Multiple bands
AUC (analytical ultracentrifugation): Gold standard
The numbers:
Removing N-glycosylation: 5-10× improvement in GPCR crystallization success
Ligand stabilization: 3-5× improvement (kinases, GPCRs, transporters)
Monodisperse sample (>95% monomer): 2× improvement in crystallization
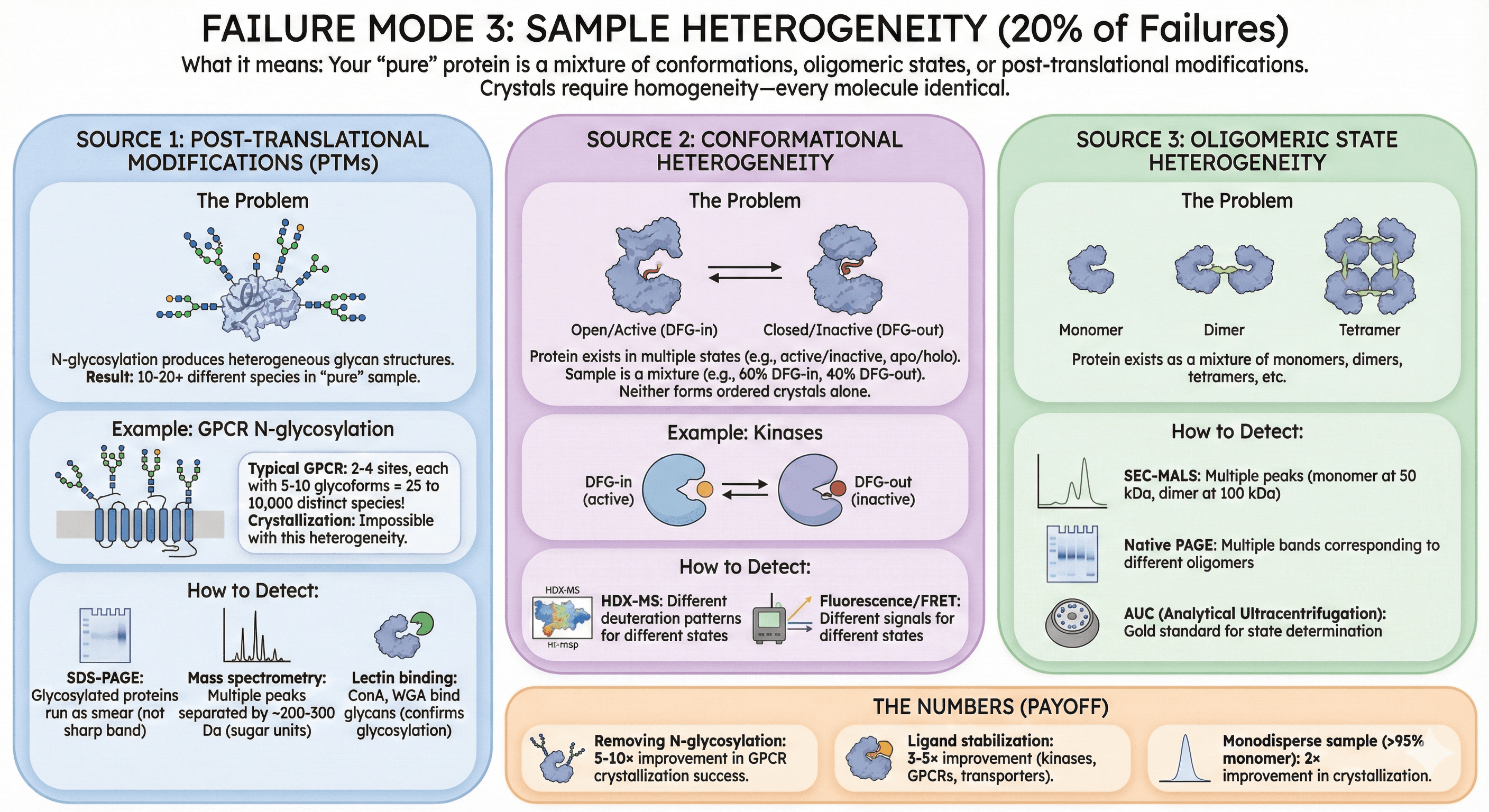
Failure Mode 4: Wrong Construct Boundaries (15% of Failures)
What it means: You've included too much (disordered regions that prevent packing) or too little (removed essential domains).
The Goldilocks Problem
Too long: Includes flexible termini or loops → prevents crystallization Too short: Removes stabilizing domains → protein unfolds or aggregates Just right: Structured core with minimal disorder
How to Diagnose
1. Check AlphaFold confidence (pLDDT):
Your construct includes regions with pLDDT < 50 (low confidence)
These disordered regions will block crystallization
Need to truncate
2. Limited proteolysis:
Native protein is proteolyzed to stable core
Run mass spec: Identify boundaries of stable fragment
This is your crystallization construct
3. Homolog comparison:
Find crystal structures of homologs in PDB
Check what boundaries they used
Often crystallized constructs are truncated versions
Common Boundary Mistakes
Including disordered N/C-termini:
Example: Your construct is residues 1-350
AlphaFold shows residues 1-25 and 320-350 are disordered (pLDDT < 50)
Better construct: residues 26-319
Removing structured domains:
Example: You truncate to "just the catalytic domain" (residues 100-250)
But residues 250-300 are a stabilizing helix
Protein without this helix aggregates
Better construct: Include 100-300
Not removing flexible loops in GPCRs:
ICL3 (intracellular loop 3): Highly flexible, 20-60 residues
Blocks crystallization (prevents ordered packing)
Solution: Replace ICL3 with T4 lysozyme (fusion protein)
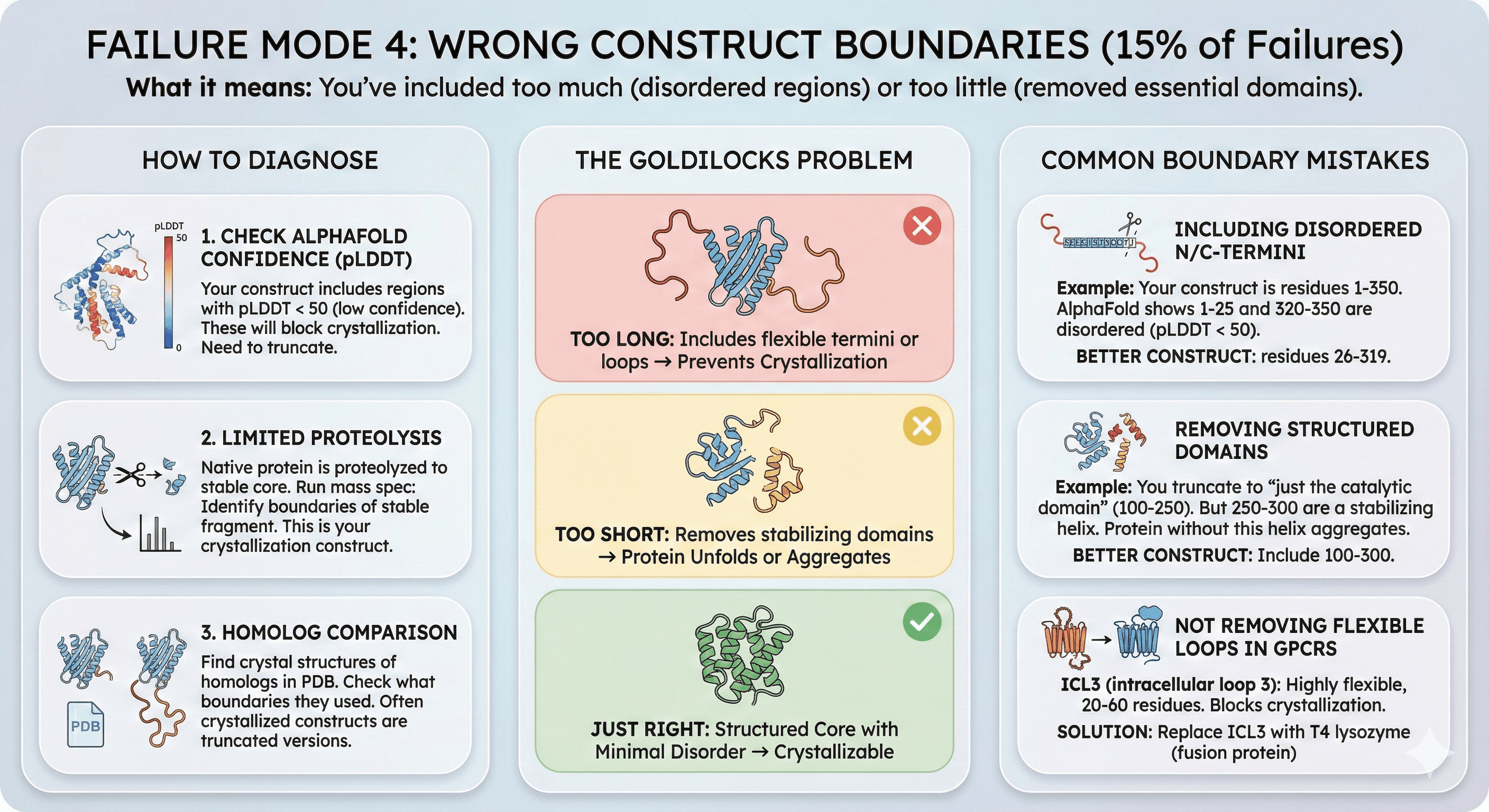
Failure Mode 5: Aggregation Propensity (20% of Failures)
What it means: Your protein has surface-exposed hydrophobic patches that cause aggregation at crystallization concentrations (5-20 mg/mL).
The Concentration Problem
During purification:
Protein at 0.5-2 mg/mL: Soluble, monodisperse
SEC-MALS looks perfect
During crystallization:
Concentrate to 10-20 mg/mL
Protein aggregates (oligomers form)
Aggregates precipitate or form amorphous aggregates (not crystals)
Why aggregation blocks crystallization:
Aggregates are heterogeneous (different sizes)
Cannot form ordered lattice
Often irreversible (once aggregated, cannot dissociate)
How to Diagnose
1. DLS (Dynamic Light Scattering):
At 1 mg/mL: Monodisperse (Rh = 3.5 nm, single peak)
At 10 mg/mL: Polydisperse (two peaks: 3.5 nm and 15 nm)
Conclusion: Concentration-dependent aggregation
2. SEC-MALS at different concentrations:
Run at 0.5, 2, 5, 10 mg/mL
If higher-molecular-weight species appear at high concentration → aggregation
3. Thermal shift assay with aggregation dye:
SYPRO Orange (standard) detects unfolding
Aggregation dyes (ProteoStat, ThT) detect aggregates
If aggregation curve appears before unfolding → aggregation-prone
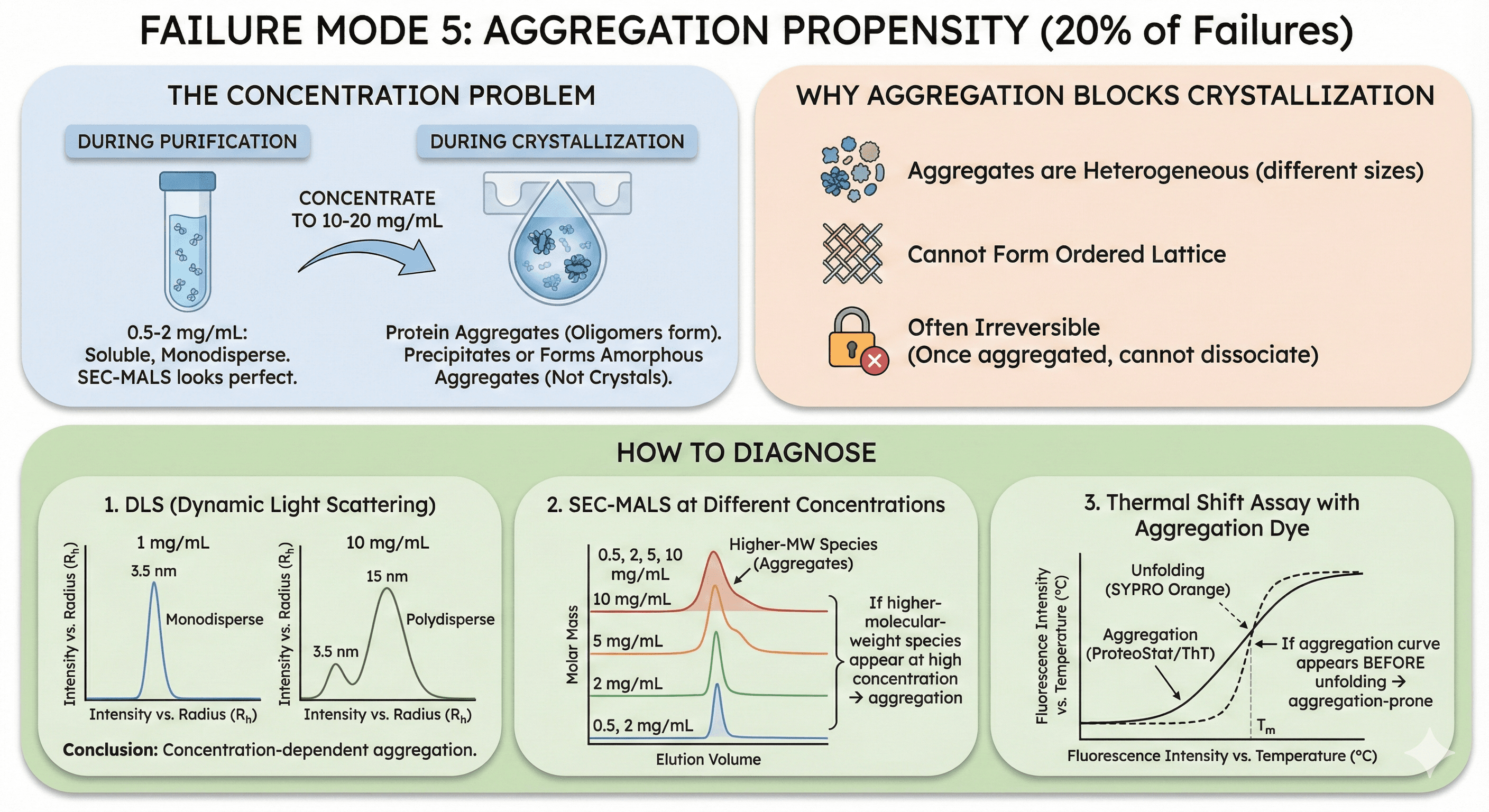
Failure Mode 6: Missing Cofactors or Binding Partners (5% of Failures)
What it means: Your protein requires a cofactor (metal ion, heme, nucleotide) or binding partner to fold correctly or stabilize. Without it, the protein is unstable or heterogeneous.
Common Missing Cofactors
Metal ions:
Zinc (Zn²⁺): Zinc finger domains, metalloproteases
Magnesium (Mg²⁺): Kinases, phosphatases, nucleic acid-binding proteins
Calcium (Ca²⁺): EF-hand domains, some proteases
Iron (Fe²⁺/Fe³⁺): Heme proteins, iron-sulfur clusters
Organic cofactors:
Heme: Cytochromes, peroxidases, hemoglobin
FAD/FMN: Flavoproteins, oxidoreductases
NAD/NADP: Dehydrogenases
ATP/ADP: Kinases, ATPases
How Cofactors Affect Crystallization
Without cofactor:
Binding pocket is "empty" and flexible
Conformational heterogeneity (multiple states)
Lower thermal stability (Tm drops 5-15°C)
Result: Won't crystallize
With cofactor:
Binding pocket occupied and rigid
Homogeneous conformation
Stabilized structure
Result: Crystals form
How to Diagnose
1. Predict cofactor requirements:
Check UniProt: Known cofactors for homologs
Literature: What cofactors do family members use?
Orbion: Predicts metal-binding sites, cofactor requirements
2. Thermal shift with cofactor:
Measure Tm without cofactor: 52°C
Measure Tm with Zn²⁺: 64°C (+12°C increase)
Conclusion: Zinc is required for stability
3. Activity assays:
If enzyme, measure activity ± cofactor
If no activity without cofactor → it's essential
Case Study: Kinase Crystallization
Target: Novel kinase, full-length
Attempt 1: Apo kinase (no ligands)
Purification: Monodisperse
Tm: 48°C (low)
Crystallization: No crystals (6 months, 2,000 conditions)
Attempt 2: Kinase + Mg²⁺ + ATP analog (AMP-PNP)
Add 5 mM MgCl₂ and 2 mM AMP-PNP during purification
Tm: 58°C (+10°C)
Crystallization: Crystals in 3 weeks
Diffraction: 2.4 Å resolution
Lesson: Many enzymes absolutely require cofactors/ligands for crystallization.
Understanding Your Failure Mode: Quick Diagnostic
Before you set up 2,000 more crystallization conditions, determine which failure mode you're facing:
Symptom | Likely Failure Mode | Quick Test |
|---|---|---|
AlphaFold has long orange/red regions | Flexibility | Check pLDDT plot |
High % of surface Lys/Glu/Arg | Surface entropy | Calculate surface composition |
SDS-PAGE shows smear (not band) | PTM heterogeneity | Mass spec |
SEC-MALS shows multiple peaks | Oligomeric heterogeneity | AUC or native PAGE |
Construct includes disordered termini | Wrong boundaries | Compare to homolog structures |
Aggregates at >5 mg/mL | Aggregation | DLS at multiple concentrations |
Low Tm (<50°C) | Missing cofactors | Thermal shift ± cofactors |
Key Takeaway
Crystallization failure isn't random bad luck. It's a predictable engineering problem with known failure modes:
Flexibility (35%): Disordered regions prevent packing
Surface entropy (25%): Flexible surface residues oppose crystallization
Heterogeneity (20%): PTMs, conformational states, oligomers
Wrong boundaries (15%): Too much or too little protein
Aggregation (20%): Concentration-dependent oligomerization
Missing cofactors (5%): Protein unstable without ligands
Understanding your failure mode is the first step to solving it.
Book a 20-Minute Demo
Sign up free for unlimited Overview runs — summary, sequence-based analysis, homology search. For the full Characterization — PTMs, binding sites, stability variants, construct design — book a demo and we'll run your target live.
