Blog
Orbion Team
How to Fix Crystallization Problems: The Modern Troubleshooting Guide

There are 6 common reasons why proteins won't crystallize: flexibility, surface entropy, heterogeneity, wrong boundaries, aggregation, and missing cofactors. The critical question is that: How do you fix it?
We'll cover systematic solutions for each failure mode, modern AI-driven prediction, and a real-world case study of rescuing an "uncrystallizable" GPCR.
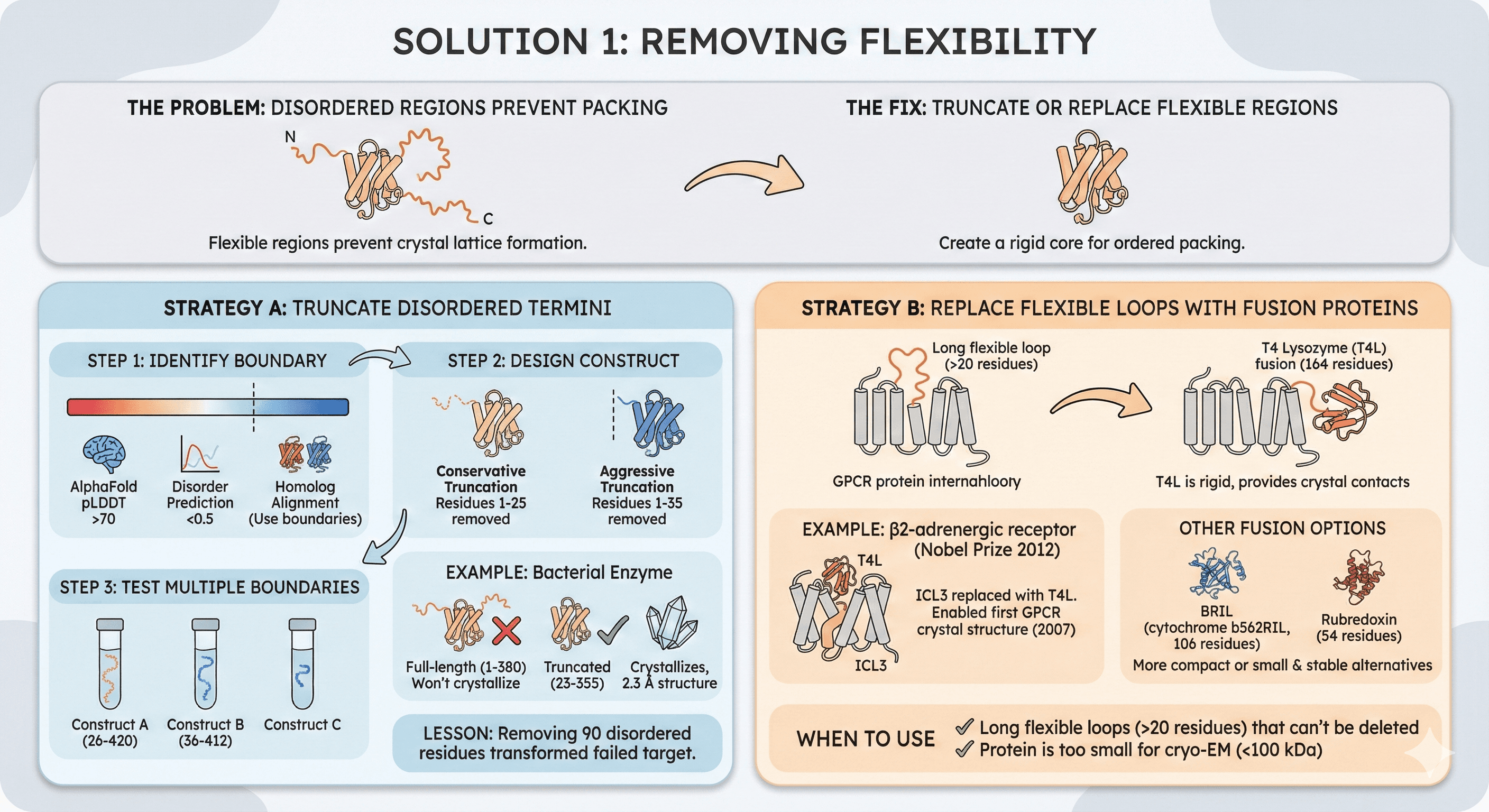
Solution 1: Removing Flexibility
The problem: Disordered termini or flexible loops prevent crystal packing.
The fix: Truncate or replace flexible regions.
Strategy A: Truncate Disordered Termini
Step 1: Identify boundary
AlphaFold pLDDT: First/last residue with pLDDT >70
Disorder prediction (IUPred): First/last residue with score <0.5
Homolog alignment: Use boundaries from closest crystal structure
Step 2: Design construct
Conservative truncation: Remove only clearly disordered regions
Example: If residues 1-25 have pLDDT <50, start at residue 26
Aggressive truncation: Remove all questionable regions
Example: If residues 1-35 have pLDDT <70, start at residue 36
Step 3: Test multiple boundaries
Design 2-3 constructs with different boundaries
Construct A: Residues 26-420 (conservative)
Construct B: Residues 36-412 (aggressive)
Example: Bacterial enzyme
Full-length (1-380): Won't crystallize
Truncated (23-355): Crystallizes, 2.3 Å structure
Lesson: Removing 90 disordered residues transformed failed target
Strategy B: Replace Flexible Loops with Fusion Proteins
For GPCRs and proteins with long internal loops (>20 residues)
T4 Lysozyme (T4L) fusion:
Replace flexible loop (e.g., GPCR ICL3) with T4 lysozyme (164 residues)
T4L is rigid, provides crystal contacts
Example: β2-adrenergic receptor (Nobel Prize 2012)
ICL3 replaced with T4L
Enabled first GPCR crystal structure (2007)
Other fusion options:
BRIL (cytochrome b562RIL): 106 residues, more compact than T4L
Rubredoxin: 54 residues, small and stable
When to use:
Long flexible loops (>20 residues) that can't be deleted
Protein is too small for cryo-EM (<100 kDa)
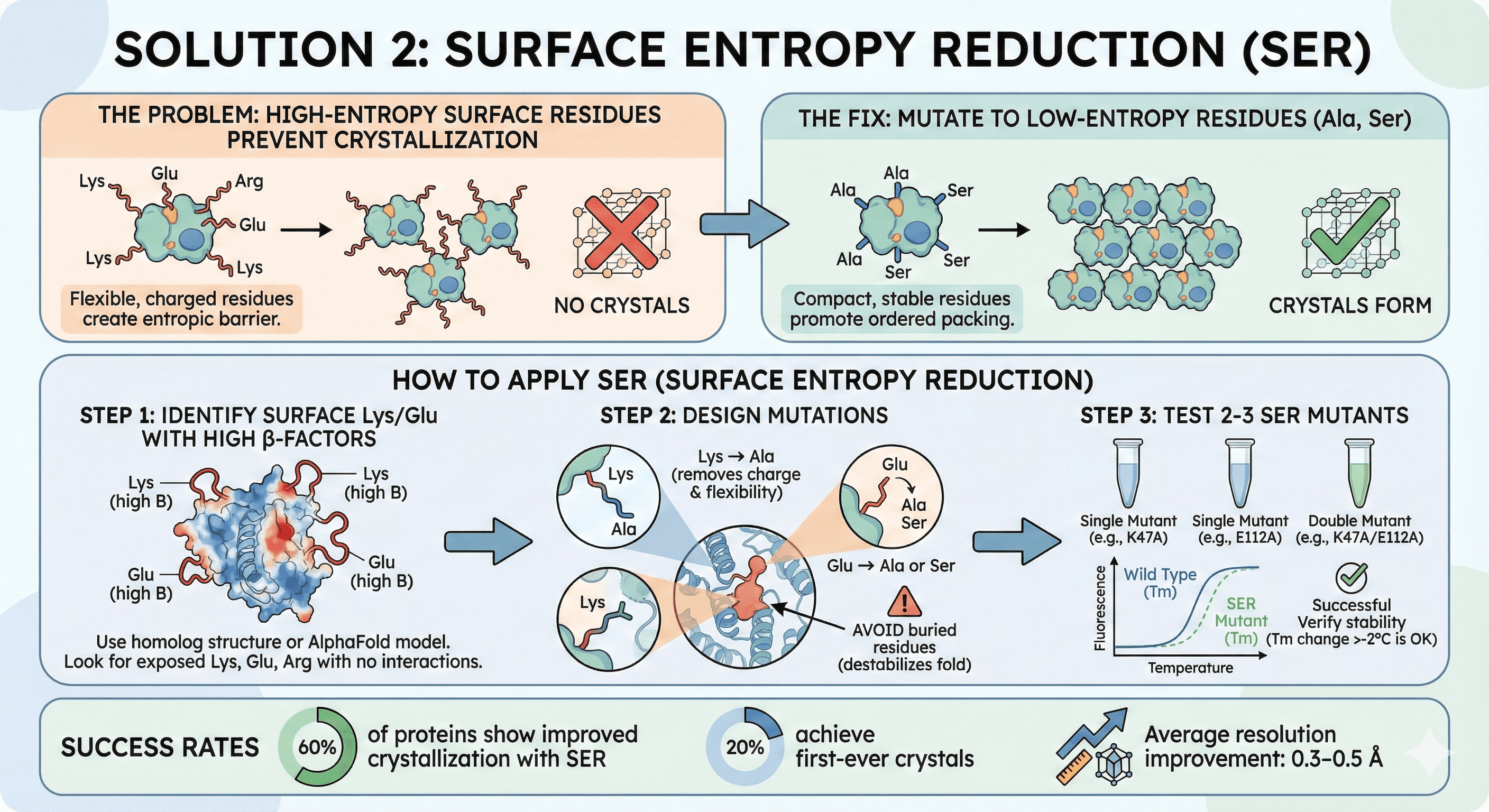
Solution 2: Surface Entropy Reduction (SER)
The problem: High-entropy surface residues (Lys, Glu, Arg) prevent crystallization.
The fix: Mutate to low-entropy residues (Ala, Ser).
How to Apply SER
Step 1: Identify surface Lys/Glu with high B-factors
Use structure of homolog or AlphaFold model
Look for Lys, Glu, Arg on surface with no obvious interactions
Step 2: Design mutations
Lys → Ala (removes charge and flexibility)
Glu → Ala or Ser
Avoid buried residues (destabilizes fold)
Step 3: Test 2-3 SER mutants
Single mutations: K47A, E112A
Double mutant: K47A/E112A
Verify stability (Tm should not decrease >2°C)
Success rates:
60% of proteins show improved crystallization with SER
20% achieve first-ever crystals
Average resolution improvement: 0.3-0.5 Å
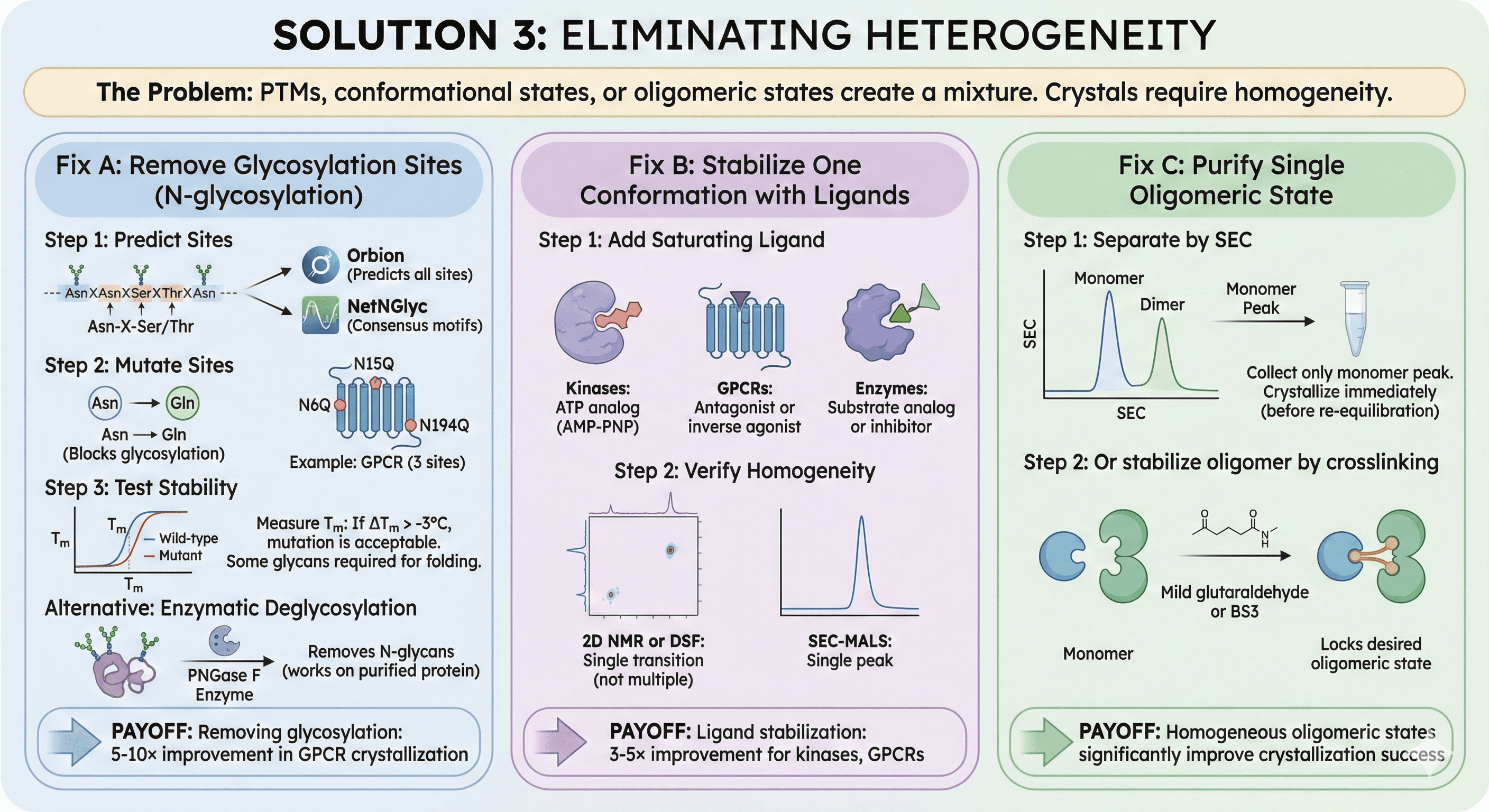
Solution 3: Eliminating Heterogeneity
The problem: PTMs, conformational states, or oligomeric states create a mixture.
Fix A: Remove Glycosylation Sites
For N-glycosylation (Asn-X-Ser/Thr motifs):
Step 1: Predict sites
Use Orbion (predicts all glycosylation sites)
Or NetNGlyc (free, consensus motifs only)
Step 2: Mutate sites
Asn → Gln (blocks glycosylation, conservative mutation)
Example: GPCR with 3 sites (N6Q, N15Q, N194Q)
Step 3: Test stability
Some glycans are required for folding
Measure Tm: If ΔTm > -3°C, mutation is acceptable
Alternative: Enzymatic deglycosylation
PNGase F: Removes N-glycans (works on purified protein)
Treat protein, then crystallize deglycosylated form
Impact:
Removing glycosylation: 5-10× improvement in GPCR crystallization
Fix B: Stabilize One Conformation with Ligands
For proteins in multiple conformational states:
Step 1: Add saturating ligand
Kinases: ATP analog (AMP-PNP)
GPCRs: Antagonist or inverse agonist
Enzymes: Substrate analog or inhibitor
Step 2: Verify homogeneity
2D NMR or DSF: Should see single transition (not multiple)
SEC-MALS: Single peak
Impact:
Ligand stabilization: 3-5× improvement for kinases, GPCRs
Fix C: Purify Single Oligomeric State
For proteins in monomer-dimer equilibrium:
Step 1: Separate by SEC
Collect only monomer peak
Crystallize immediately (before re-equilibration)
Step 2: Or stabilize oligomer by crosslinking
Mild glutaraldehyde or BS3
Locks desired oligomeric state
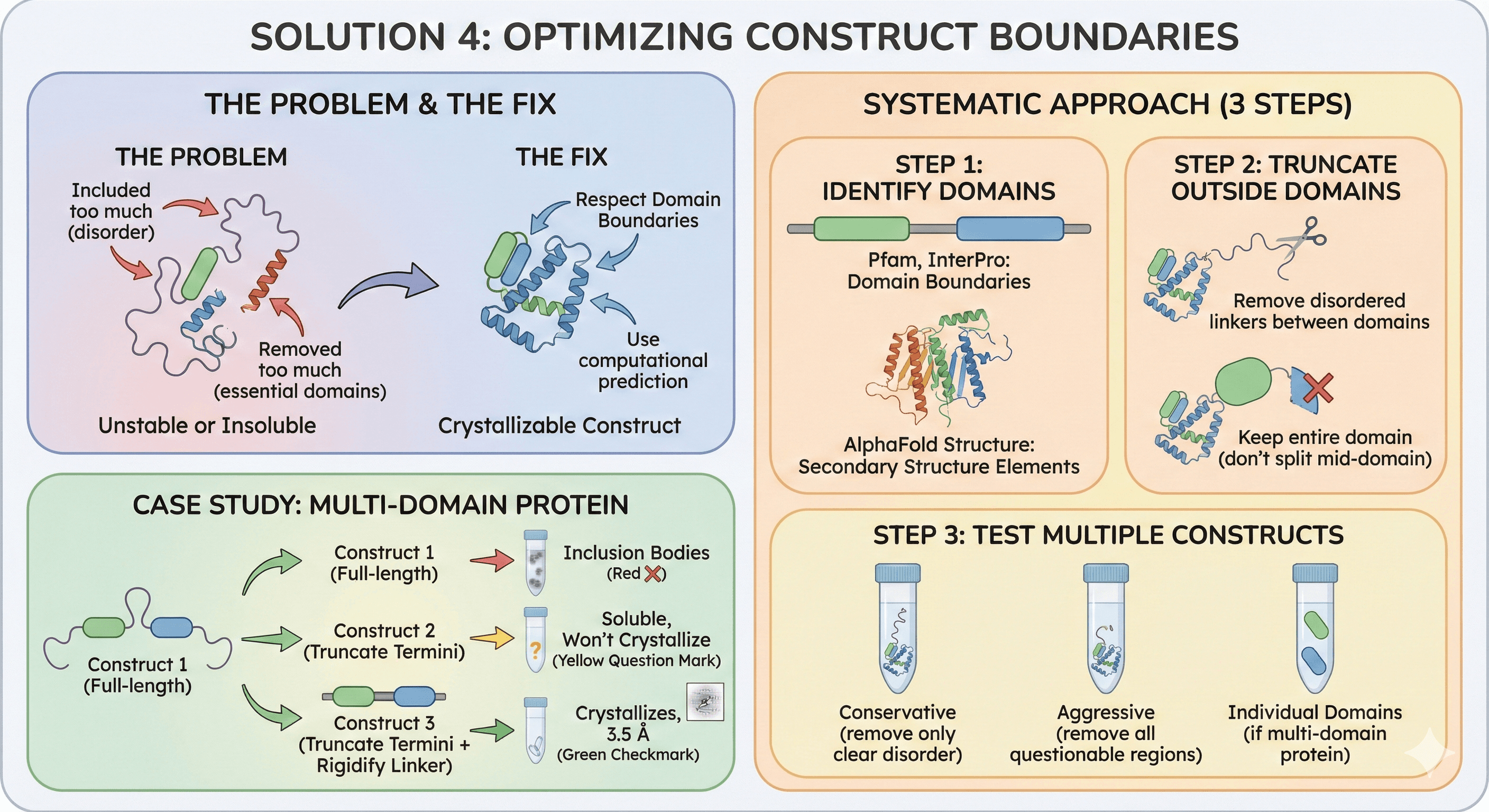
Solution 4: Optimizing Construct Boundaries
The problem: Included too much (disorder) or removed too much (essential domains).
The fix: Respect domain boundaries, use computational prediction.
Systematic Approach
Step 1: Identify domains
Pfam, InterPro: Domain boundaries
AlphaFold structure: Secondary structure elements
Step 2: Truncate outside domains
Remove disordered linkers between domains
Keep entire domain (don't split mid-domain)
Step 3: Test multiple constructs
Conservative (remove only clear disorder)
Aggressive (remove all questionable regions)
Individual domains (if multi-domain protein)
Case study: Multi-domain protein
Construct 1 (full-length): Inclusion bodies
Construct 2 (truncate termini): Soluble, won't crystallize
Construct 3 (truncate termini + rigidify linker): Crystallizes, 3.5 Å
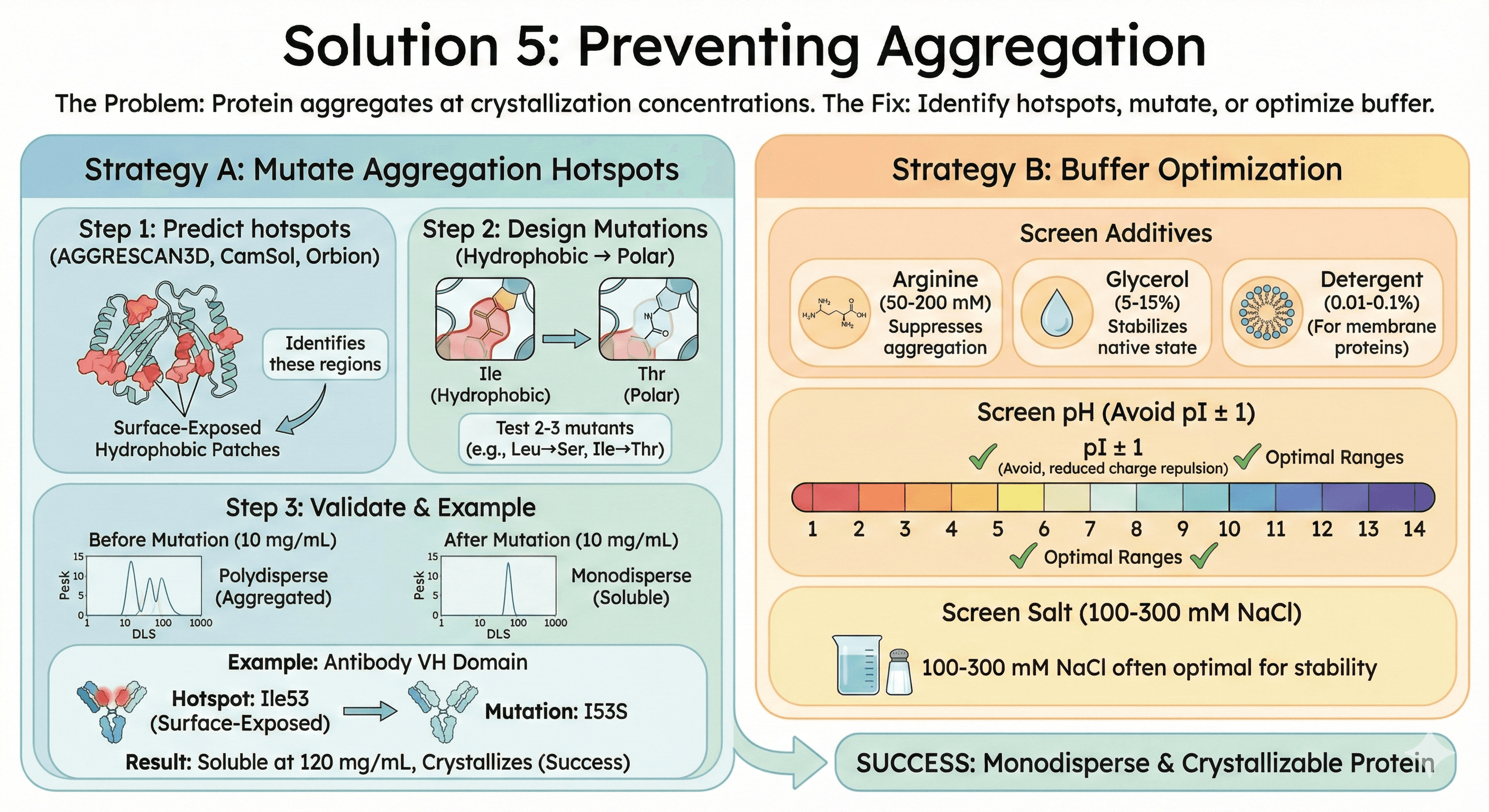
Solution 5: Preventing Aggregation
The problem: Protein aggregates at crystallization concentrations.
The fix: Identify hotspots, mutate, or optimize buffer.
Strategy A: Mutate Aggregation Hotspots
Step 1: Predict hotspots
AGGRESCAN3D, CamSol, or Orbion
Identifies surface-exposed hydrophobic patches
Step 2: Design mutations
Hydrophobic → Polar: Leu→Ser, Ile→Thr
Test 2-3 mutants
Step 3: Validate
DLS at 10 mg/mL: Should be monodisperse
Crystallization trials
Example: Antibody VH domain
Problem: Aggregates above 50 mg/mL
Hotspot: Ile53 in CDR2 (surface-exposed)
Mutation: I53S
Result: Soluble at 120 mg/mL, crystallizes
Strategy B: Buffer Optimization
Screen additives:
Arginine (50-200 mM): Suppresses aggregation
Glycerol (5-15%): Stabilizes native state
Detergent (0.01-0.1% for membrane proteins)
Screen pH:
Avoid pI ± 1 pH unit (reduced charge repulsion)
Screen salt:
100-300 mM NaCl often optimal
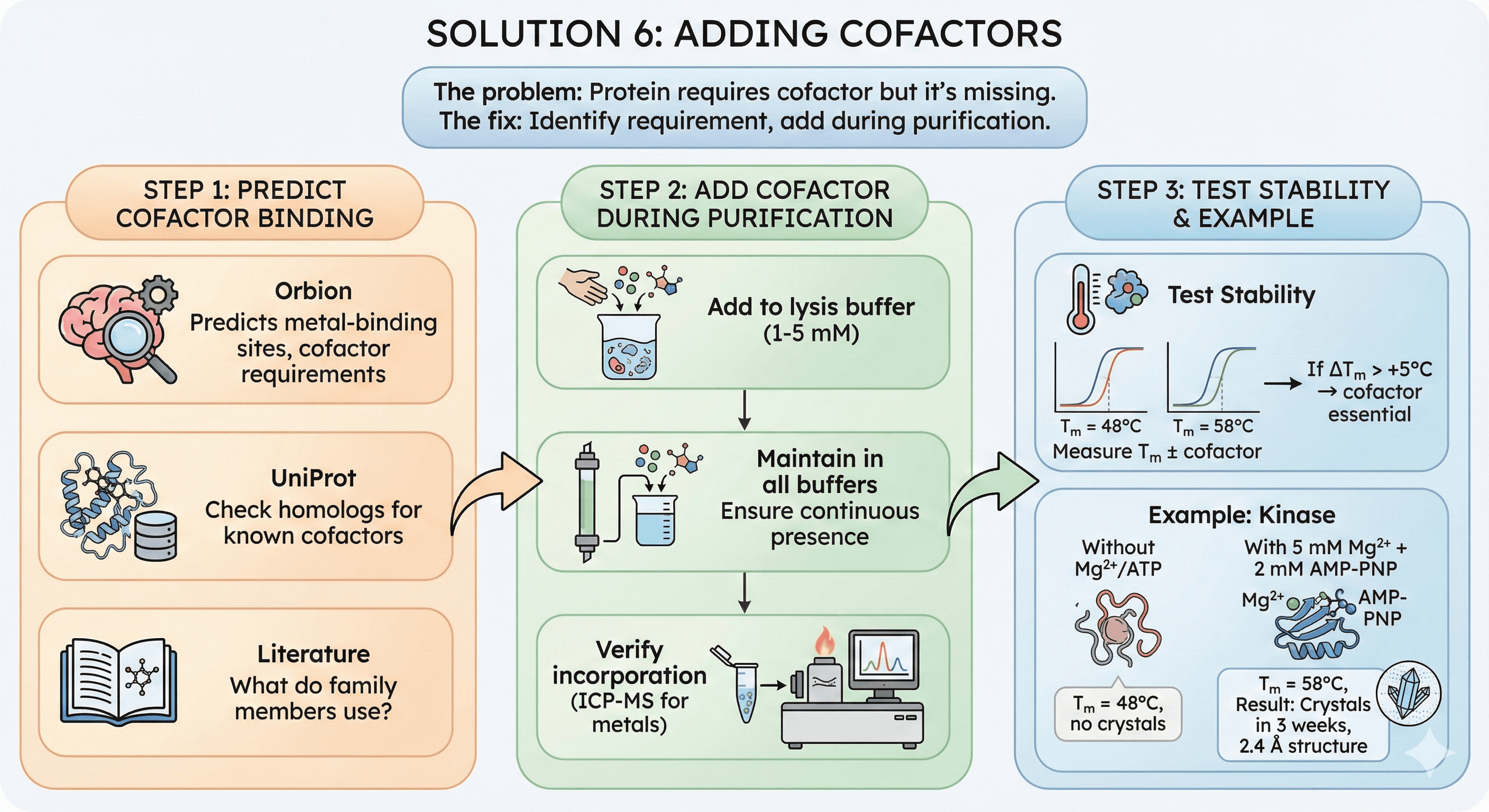
Solution 6: Adding Cofactors
The problem: Protein requires cofactor but it's missing.
The fix: Identify requirement, add during purification.
How to Fix
Step 1: Predict cofactor binding
Orbion: Predicts metal-binding sites, cofactor requirements
UniProt: Check homologs
Literature: What do family members use?
Step 2: Add cofactor during purification
Add to lysis buffer (1-5 mM)
Maintain in all buffers
Verify incorporation (ICP-MS for metals)
Step 3: Test stability
Measure Tm ± cofactor
If ΔTm > +5°C → cofactor essential
Example: Kinase
Without Mg²⁺/ATP: Tm = 48°C, no crystals
With 5 mM Mg²⁺ + 2 mM AMP-PNP: Tm = 58°C
Result: Crystals in 3 weeks, 2.4 Å structure
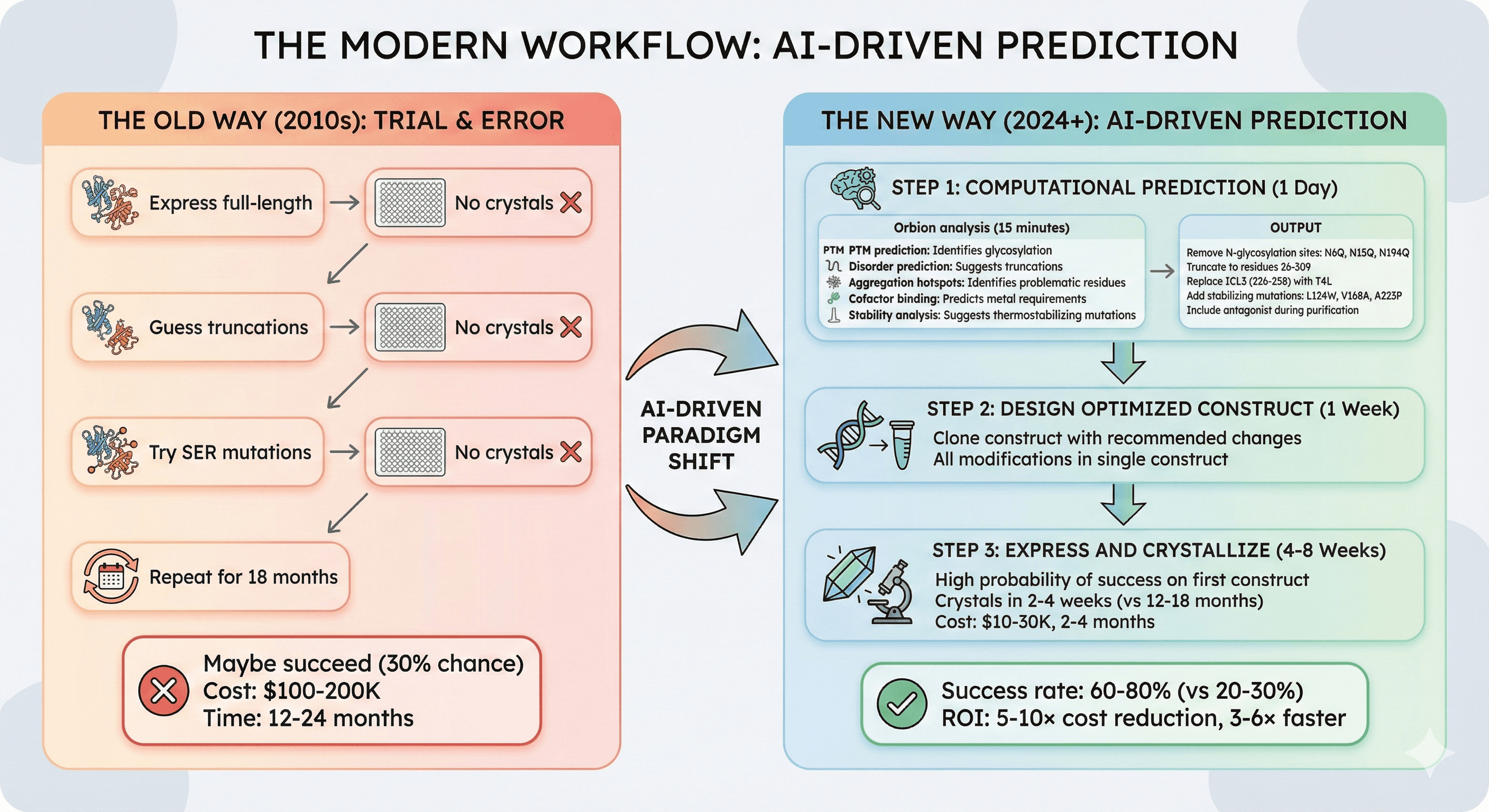
The Modern Workflow: AI-Driven Prediction
The old way (2010s):
Express full-length → No crystals
Guess truncations → No crystals
Try SER mutations → No crystals
Repeat for 18 months
Maybe succeed (30% chance)
Cost: $100-200K, 12-24 months
The new way (2024+):
Step 1: Computational Prediction (1 Day)
Orbion analysis (15 minutes):
PTM prediction: Identifies glycosylation causing heterogeneity
Disorder prediction: Suggests truncation boundaries
Aggregation hotspots: Identifies problematic residues
Cofactor binding: Predicts metal requirements
Stability analysis: Suggests thermostabilizing mutations
Output:
"Remove N-glycosylation sites: N6Q, N15Q, N194Q"
"Truncate to residues 26-309"
"Replace ICL3 (residues 226-258) with T4L"
"Add stabilizing mutations: L124W, V168A, A223P"
"Include antagonist during purification"
Step 2: Design Optimized Construct (1 Week)
Clone construct with recommended changes
All modifications in single construct
Step 3: Express and Crystallize (4-8 Weeks)
High probability of success on first construct
Crystals in 2-4 weeks (vs 12-18 months traditional)
Cost: $10-30K, 2-4 months
Success rate: 60-80% (vs 20-30% traditional)
ROI: 5-10× cost reduction, 3-6× faster
Case Study: Rescuing an "Uncrystallizable" GPCR
The Challenge
Target: Orphan GPCR (therapeutic target)
Traditional attempts (2015-2017):
Construct: Full-length (1-348)
Expression: Sf9 insect cells, low yield (0.5 mg/L)
Tm: 42°C (very unstable)
Crystallization: 3,000+ conditions over 18 months, no crystals
Cost: $250K, 2 years
Result: Project shelved as "uncrystallizable"
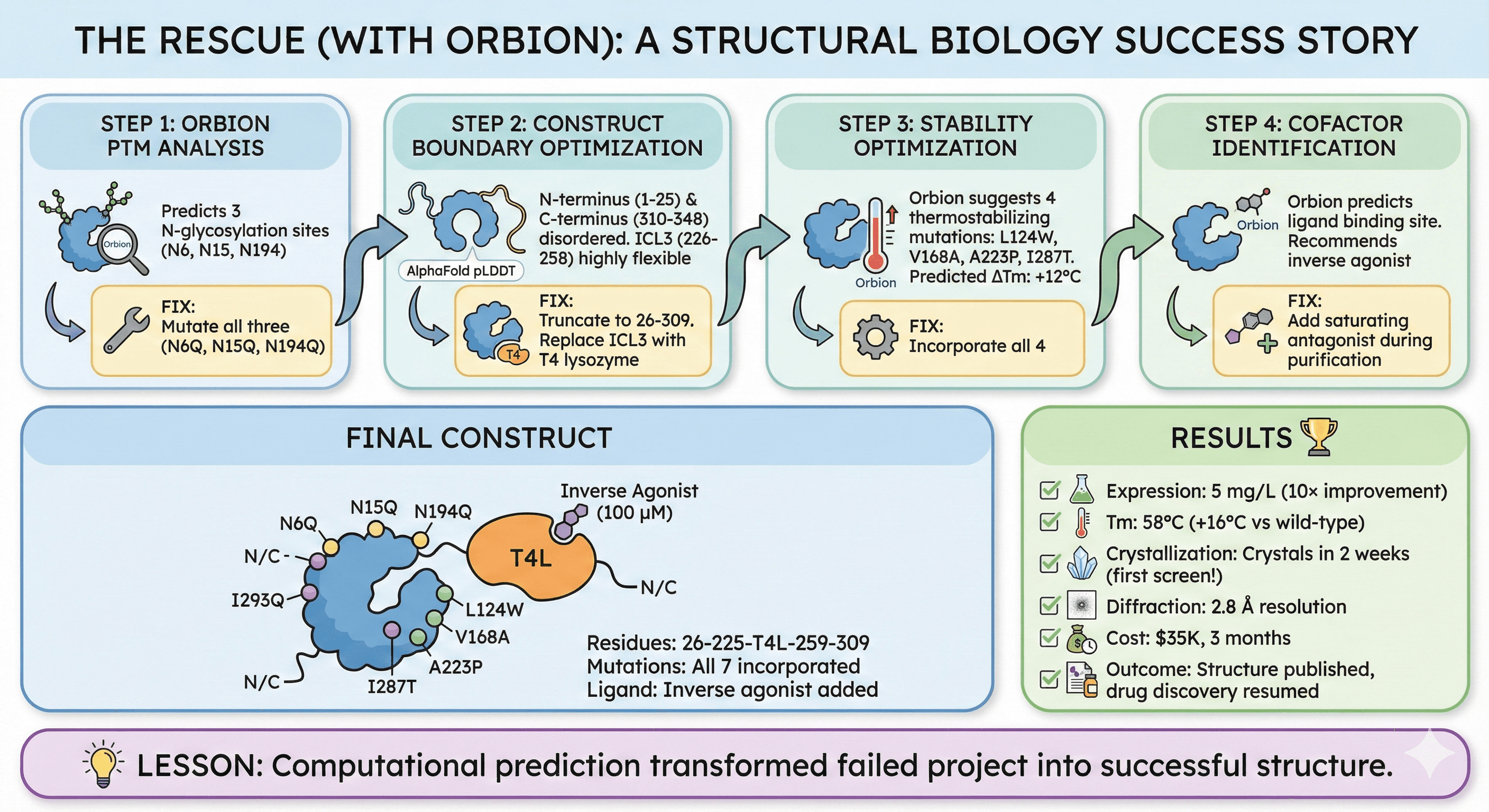
The Rescue (with Orbion)
Step 1: Orbion PTM analysis
Predicts 3 N-glycosylation sites (N6, N15, N194)
Fix: Mutate all three (N6Q, N15Q, N194Q)
Step 2: Construct boundary optimization
AlphaFold pLDDT: N-terminus (1-25) and C-terminus (310-348) disordered
ICL3 (226-258) highly flexible
Fix:
Truncate to 26-309
Replace ICL3 with T4 lysozyme
Step 3: Stability optimization
Orbion suggests 4 thermostabilizing mutations:
L124W, V168A, A223P, I287T
Predicted ΔTm: +12°C
Fix: Incorporate all 4
Step 4: Cofactor identification
Orbion predicts ligand binding site
Recommends inverse agonist
Fix: Add saturating antagonist during purification
Final Construct
Residues: 26-225-T4L-259-309
Mutations: N6Q, N15Q, N194Q, L124W, V168A, A223P, I287T
Ligand: Inverse agonist (100 μM)
Results
Expression: 5 mg/L (10× improvement)
Tm: 58°C (+16°C vs wild-type)
Crystallization: Crystals in 2 weeks (first screen!)
Diffraction: 2.8 Å resolution
Cost: $35K, 3 months
Outcome: Structure published, drug discovery resumed
Lesson: Computational prediction transformed failed project into successful structure.
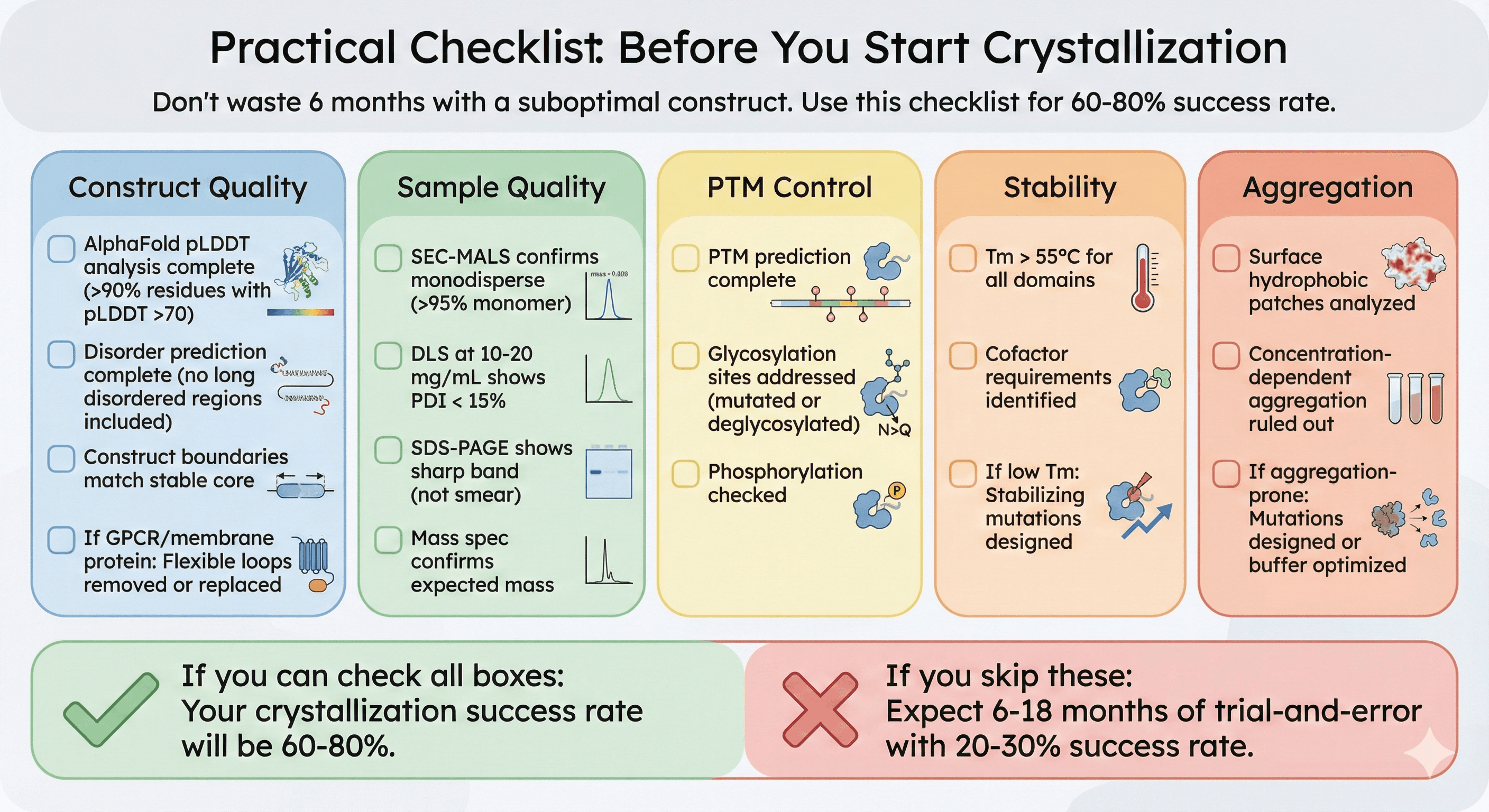
Practical Checklist: Before You Start Crystallization
Don't waste 6 months setting up screens with a suboptimal construct. Use this checklist:
☐ Construct Quality
[ ] AlphaFold pLDDT analysis complete (>90% residues with pLDDT >70)
[ ] Disorder prediction complete (no long disordered regions included)
[ ] Construct boundaries match stable core
[ ] If GPCR/membrane protein: Flexible loops removed or replaced
☐ Sample Quality
[ ] SEC-MALS confirms monodisperse (>95% monomer)
[ ] DLS at 10-20 mg/mL shows PDI < 15%
[ ] SDS-PAGE shows sharp band (not smear)
[ ] Mass spec confirms expected mass
☐ PTM Control
[ ] PTM prediction complete
[ ] Glycosylation sites addressed (mutated or deglycosylated)
[ ] Phosphorylation checked
☐ Stability
[ ] Tm > 55°C for all domains
[ ] Cofactor requirements identified
[ ] If low Tm: Stabilizing mutations designed
☐ Aggregation
[ ] Surface hydrophobic patches analyzed
[ ] Concentration-dependent aggregation ruled out
[ ] If aggregation-prone: Mutations designed or buffer optimized
If you can check all boxes: Your crystallization success rate will be 60-80%.
If you skip these: Expect 6-18 months of trial-and-error with 20-30% success rate.
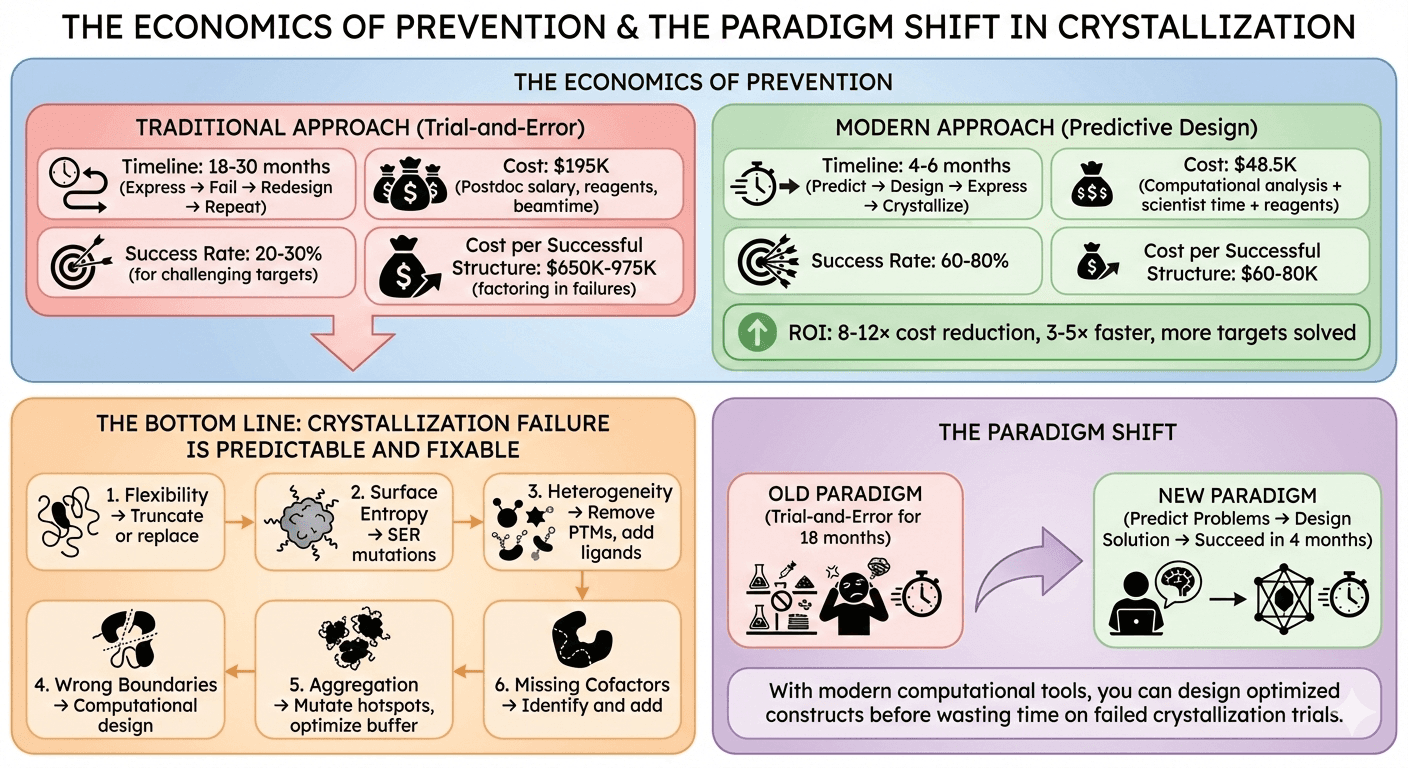
The Economics of Prevention
Traditional Approach
Timeline: 18-30 months (express → fail → redesign → repeat) Cost: $195K (postdoc salary, reagents, beamtime) Success rate: 20-30% for challenging targets Cost per successful structure: $650K-975K (factoring in failures)
Modern Approach
Timeline: 4-6 months (predict → design → express → crystallize) Cost: $48.5K (computational analysis + scientist time + reagents) Success rate: 60-80% Cost per successful structure: $60-80K
ROI: 8-12× cost reduction, 3-5× faster, more targets solved
The Bottom Line
Crystallization failure is predictable and fixable.
The 6 failure modes:
Flexibility → Truncate or replace
Surface entropy → SER mutations
Heterogeneity → Remove PTMs, add ligands
Wrong boundaries → Computational design
Aggregation → Mutate hotspots, optimize buffer
Missing cofactors → Identify and add
The paradigm shift:
Old: Trial-and-error for 18 months
New: Predict problems → Design solution → Succeed in 4 months
With modern computational tools, you can design optimized constructs before wasting time on failed crystallization trials.
Ready to Optimize Your Crystallization Construct?
If your protein won't crystallize, Orbion can identify why and suggest fixes—in minutes, not months.
Orbion provides:
Complete PTM landscape (identify glycosylation causing heterogeneity)
Construct boundary recommendations (optimal truncation points)
Cofactor binding prediction (what's missing?)
Aggregation hotspot identification with mutations
Stability optimization (thermostabilizing mutations)
From sequence to optimized construct design in <1 day.
Book a 20-Minute Demo
Sign up free for unlimited Overview runs — summary, sequence-based analysis, homology search. For the full Characterization — PTMs, binding sites, stability variants, construct design — book a demo and we'll run your target live.
