Blog
Orbion Team
Why Your Protein Aggregates (And How to Fix It): A Structural Biologist's Guide

You expressed your protein. It looked perfect in gel filtration. Then you concentrated it to 5 mg/mL and found a white precipitate at the bottom of the tube. Welcome to the most frustrating problem in protein science: aggregation. It kills more projects than any other technical failure, costs labs hundreds of thousands of dollars in wasted time, and is the reason 30-40% of potential drug targets are labeled "undruggable." Here's why it happens—and how to fix it.
Key Takeaways
60-80% of recombinant proteins show some aggregation during expression or purification
Main culprits: Exposed hydrophobic patches, partial unfolding, concentration-dependent oligomerization
Cost of failure: $50k-100k per failed protein target (6-12 months of wasted effort)
Solution: Computational prediction of aggregation hotspots + rational mutagenesis increases success rates 2-5×
Modern tools: AI-driven prediction (Orbion, AGGRESCAN3D, CamSol) dramatically reduces failure rates
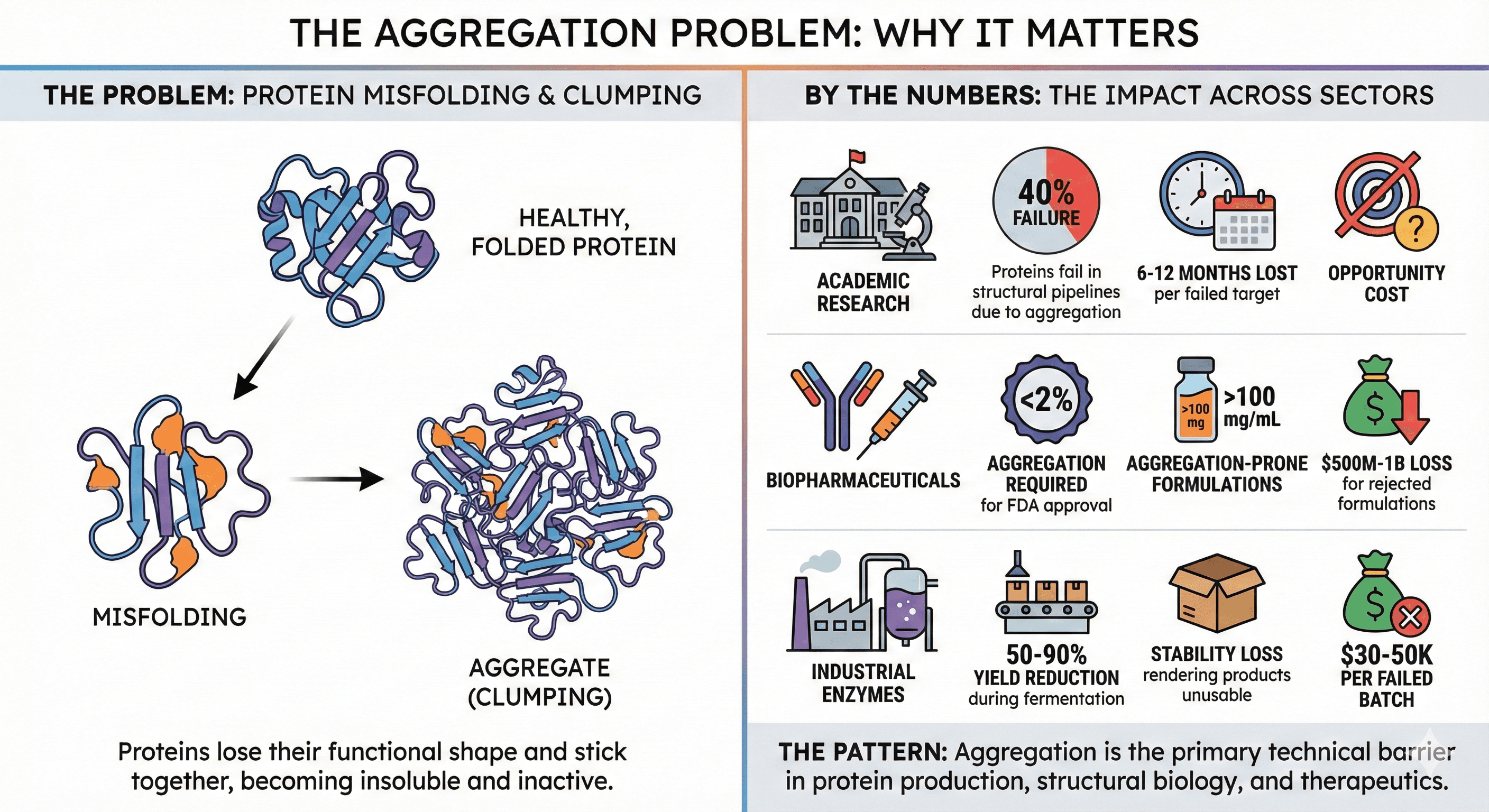
The Aggregation Problem: Why It Matters
By the Numbers
Academic research:
40% of proteins in structural genomics pipelines fail due to aggregation
Average time lost per failed target: 6-12 months
Opportunity cost: Cannot pursue alternative targets
Biopharmaceuticals:
FDA requires <2% aggregation for therapeutic antibody approval
High-concentration formulations (>100 mg/mL for subcutaneous injection) are aggregation-prone
Market impact: A rejected antibody formulation = $500M-1B loss
Industrial enzymes:
Aggregation during fermentation reduces yield 50-90%
Stability loss during storage renders products unusable
Economic impact: $30-50k per failed batch
The pattern: Aggregation isn't a niche problem. It's the primary technical barrier in protein production, structural biology, and therapeutics.
The Three Types of Aggregation
Not all aggregation is the same. Understanding the mechanism helps you choose the right fix.
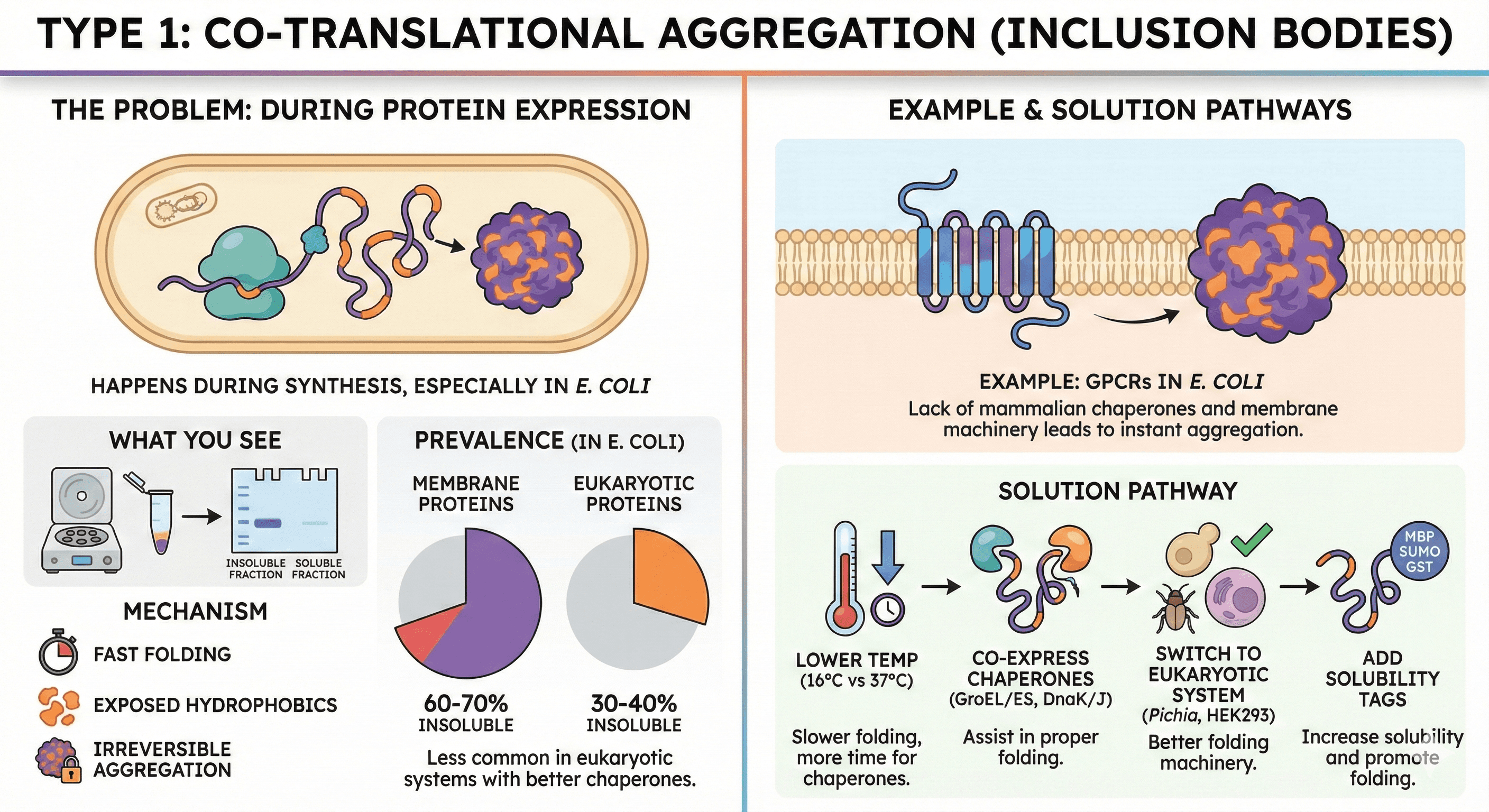
Type 1: Co-Translational Aggregation (Inclusion Bodies)
When it happens: During protein expression in the cell (usually E. coli)
What you see: Insoluble protein pellet after cell lysis. Protein is in the insoluble fraction on SDS-PAGE.
Mechanism:
Protein folds faster than chaperones can assist
Hydrophobic regions exposed during folding stick together
Misfolded intermediates aggregate irreversibly
Prevalence:
60-70% of membrane proteins in E. coli
30-40% of eukaryotic proteins in E. coli
Less common in eukaryotic expression (yeast, insect, mammalian cells have better chaperone machinery)
Example: GPCRs in E. coli G-protein coupled receptors are 7-transmembrane proteins. When expressed in bacteria:
Lack of mammalian chaperones
No membrane insertion machinery
Hydrophobic transmembrane helices aggregate instantly
Result: 100% insoluble
Solution pathway:
Try lower temperature expression (16°C vs 37°C) - slower folding, more time for chaperones
Co-express chaperones (GroEL/ES, DnaK/J)
Switch to eukaryotic system (Pichia, insect cells, HEK293)
Add solubility tags (MBP, SUMO, GST)
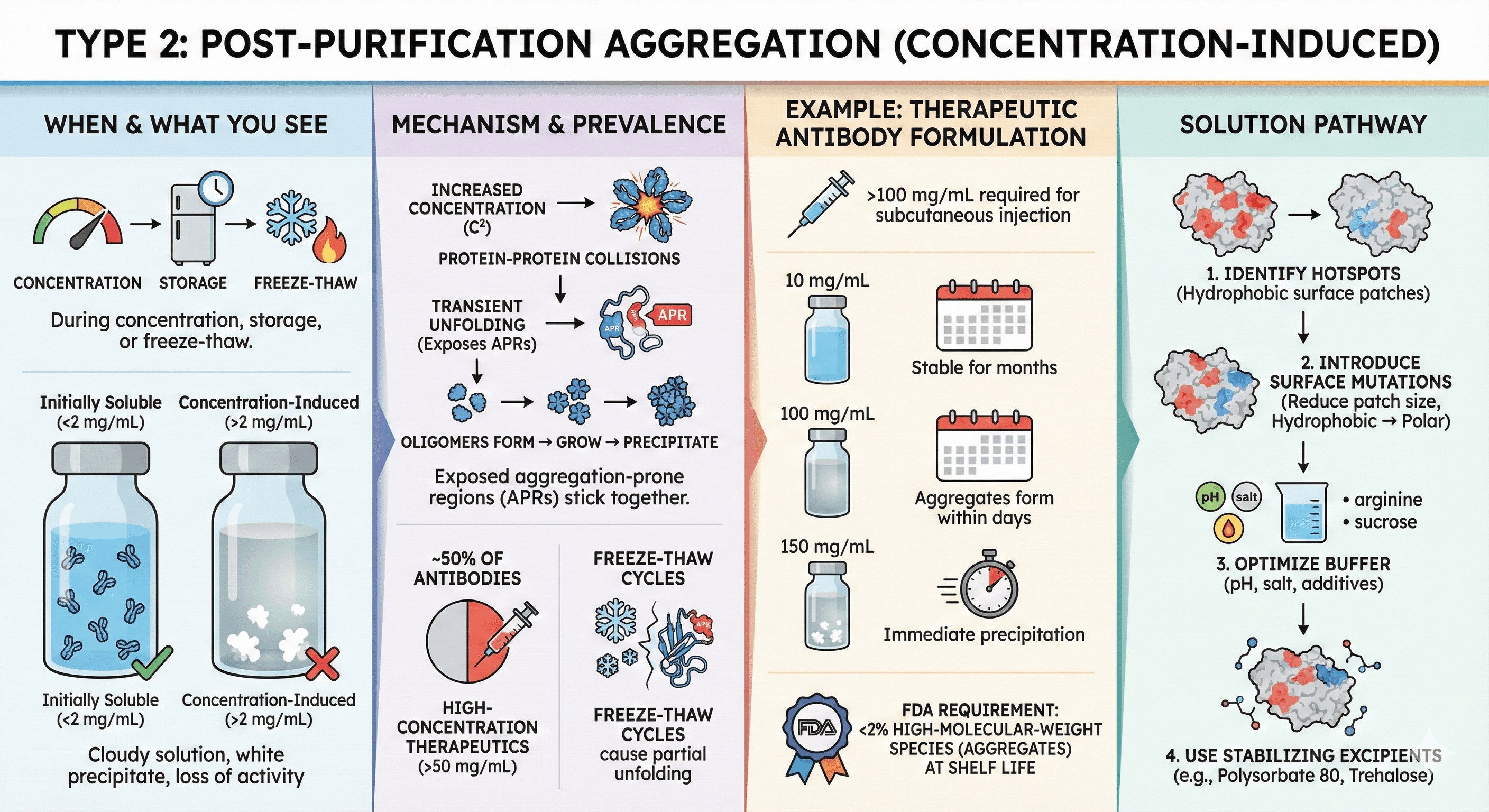
Type 2: Post-Purification Aggregation (Concentration-Induced)
When it happens: During concentration, storage, or freeze-thaw
What you see:
Protein is initially soluble after purification
Concentrating above threshold (e.g., >2 mg/mL) causes precipitation
Cloudy solution, white precipitate, loss of activity
Mechanism:
Protein-protein collisions increase with concentration (C²)
Transient unfolding exposes aggregation-prone regions (APRs)
Oligomers form → grow into larger aggregates → precipitate
Prevalence:
~50% of antibodies show concentration-dependent aggregation
Therapeutic proteins requiring high concentration (>50 mg/mL) almost always face this
Storage-induced: Freeze-thaw cycles cause partial unfolding
Example: Therapeutic Antibody Formulation Monoclonal antibodies for subcutaneous injection need >100 mg/mL:
At 10 mg/mL: Stable for months
At 100 mg/mL: Aggregates form within days
At 150 mg/mL: Immediate precipitation
FDA requirement: <2% high-molecular-weight species (aggregates) at shelf life
Solution pathway:
Identify aggregation hotspots (hydrophobic surface patches)
Introduce surface mutations to reduce patch size (hydrophobic → polar)
Optimize buffer (pH, salt, additives like arginine or sucrose)
Use stabilizing excipients (polysorbate 80, trehalose)
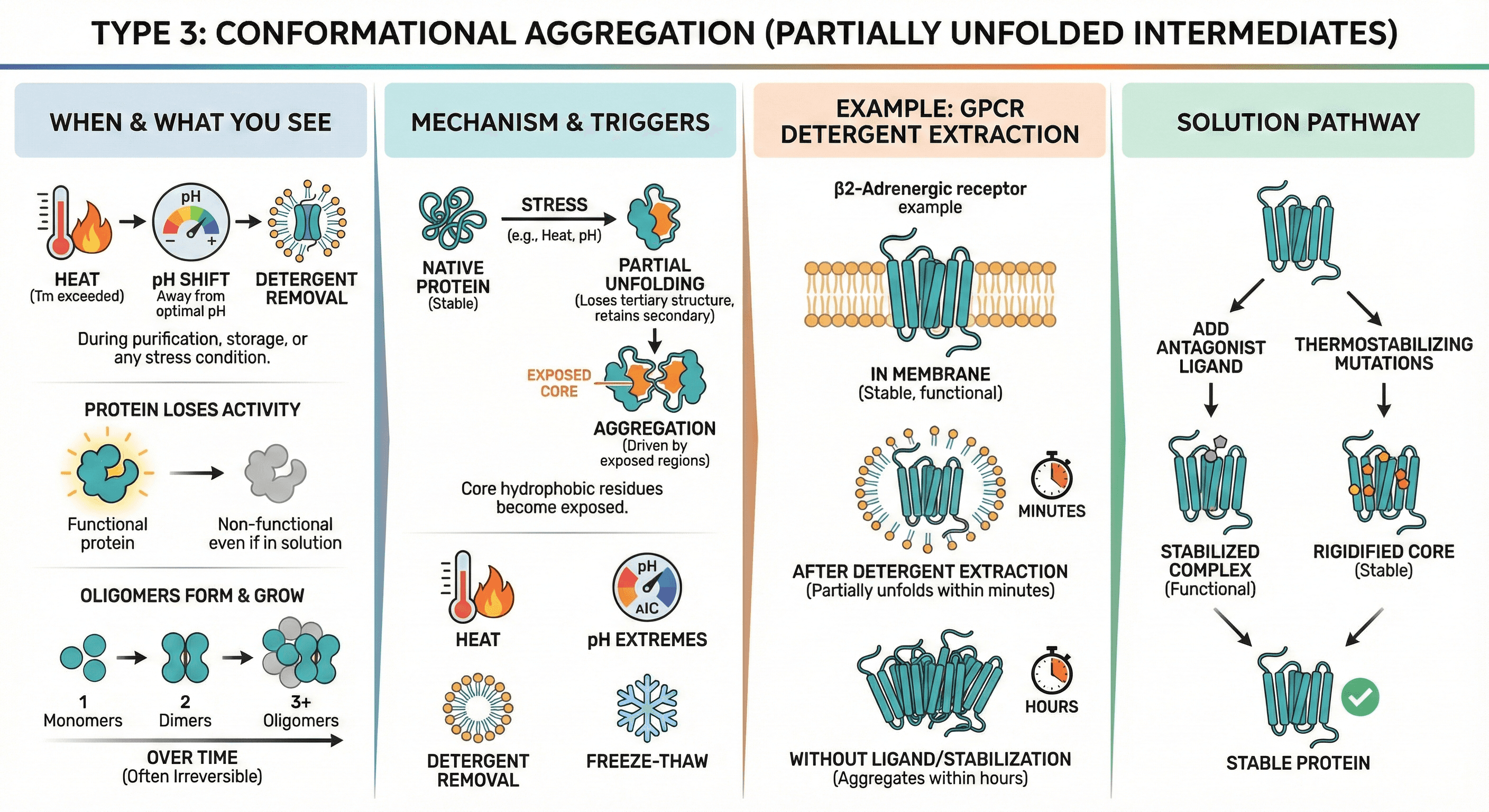
Type 3: Conformational Aggregation (Partially Unfolded Intermediates)
When it happens: During purification, storage, or any stress condition (heat, pH shift, detergent removal)
What you see:
Protein loses activity even if it stays in solution
Oligomers form (dimers, trimers) that grow over time
Often irreversible
Mechanism:
Protein partially unfolds (loses tertiary structure but retains secondary structure)
Core hydrophobic residues become exposed
These exposed regions drive aggregation
Triggers:
Heat: Tm exceeded (even briefly)
pH extremes: Away from optimal pH
Detergent removal: Membrane proteins lose stabilizing detergent
Freeze-thaw: Ice crystal formation causes local denaturation
Example: GPCR Detergent Extraction β2-Adrenergic receptor:
In membrane: Stable, functional
After detergent extraction: Partially unfolds within minutes
Without ligand/stabilization: Aggregates within hours
Solution: Add antagonist ligand (stabilizes conformation) + thermostabilizing mutations
The Molecular Basis: Why Proteins Aggregate
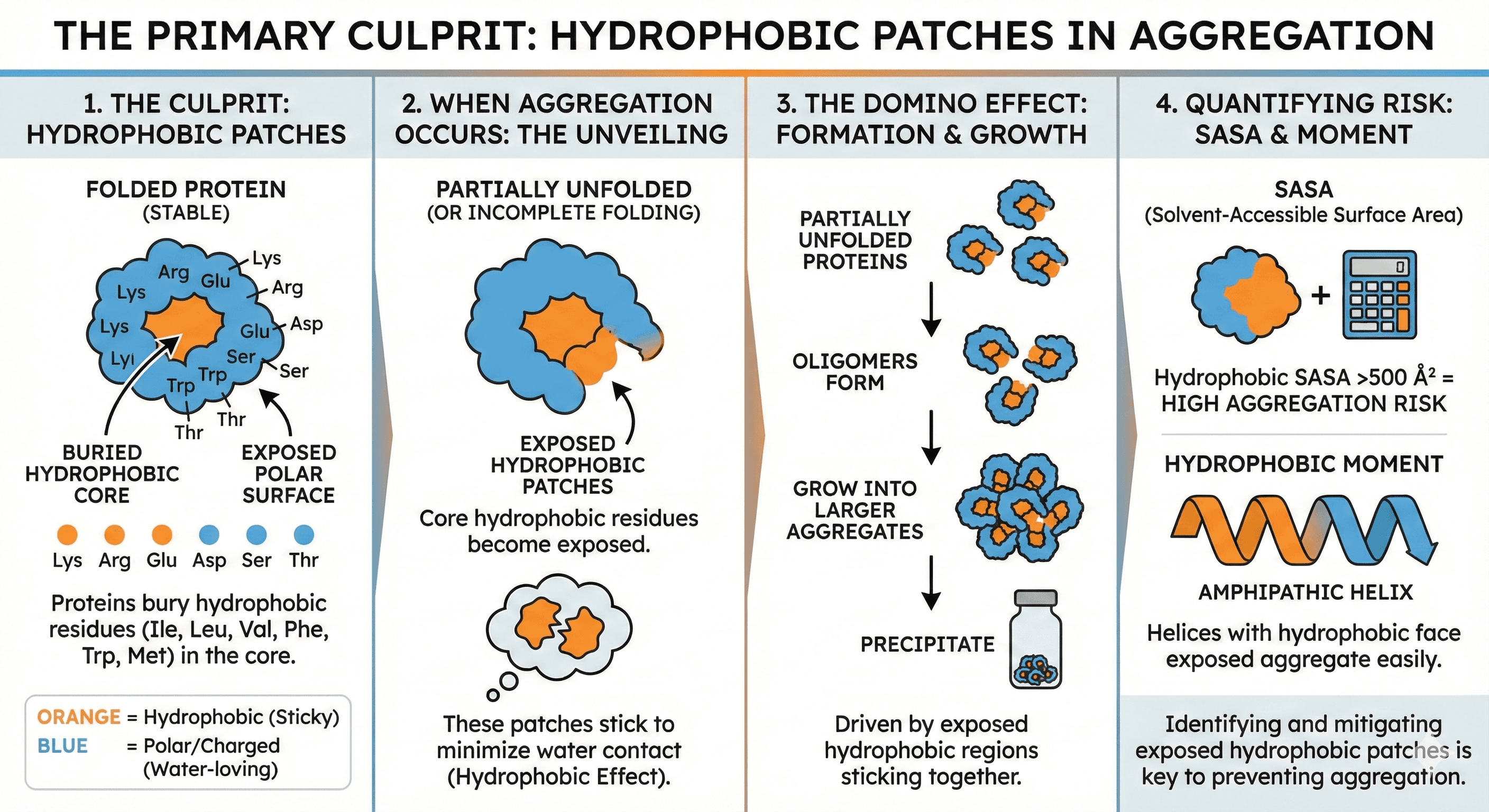
At the molecular level, aggregation is driven by one principle: Hydrophobic effect.
Hydrophobic Patches: The Primary Culprit
Proteins fold to bury hydrophobic residues (Ile, Leu, Val, Phe, Trp, Met) in the core, exposing polar/charged residues (Lys, Arg, Glu, Asp, Ser, Thr) on the surface.
When aggregation occurs:
Protein partially unfolds (or folds incompletely)
Core hydrophobic residues become exposed
These patches stick to each other to minimize water contact
Oligomers form → grow → precipitate
Quantifying exposure:
SASA (Solvent-Accessible Surface Area): Hydrophobic SASA >500 Ų = aggregation risk
Hydrophobic moment: Amphipathic helices with hydrophobic face exposed aggregate easily
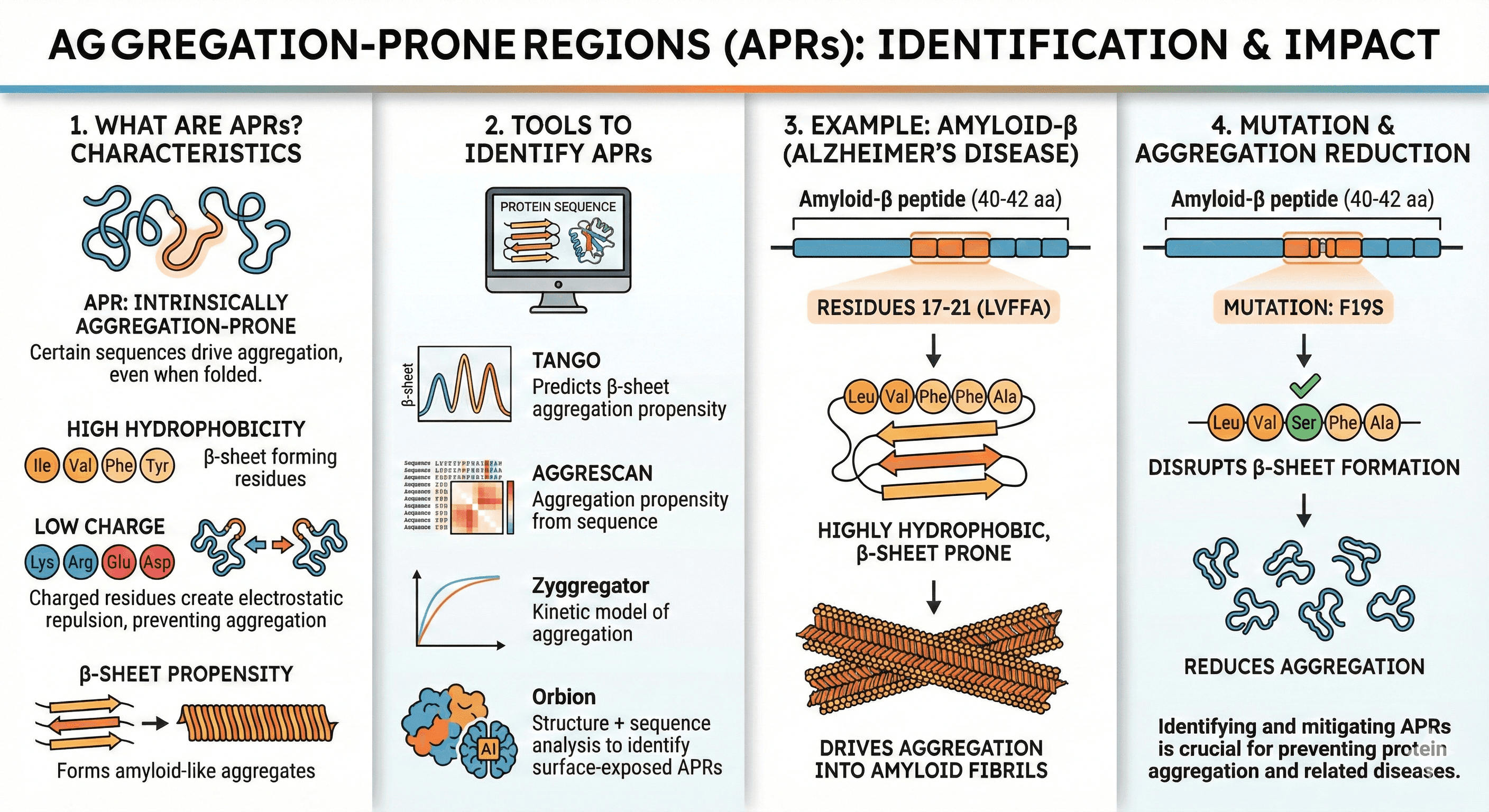
Aggregation-Prone Regions (APRs)
Certain sequences are intrinsically aggregation-prone, even in the folded state.
Characteristics of APRs:
High in hydrophobic residues (especially β-sheet forming: Ile, Val, Phe, Tyr)
Low charge (Lys, Arg, Glu, Asp prevent aggregation via electrostatic repulsion—charged residues create repulsive forces that keep protein molecules apart)
β-sheet propensity (amyloid-like aggregation)
Tools to identify APRs:
TANGO: Predicts β-sheet aggregation propensity
AGGRESCAN: Aggregation propensity from sequence
Zyggregator: Kinetic model of aggregation
Orbion: Structure + sequence analysis to identify surface-exposed APRs
Example: Amyloid-β (Alzheimer's disease) Amyloid-β peptide (40-42 amino acids):
Residues 17-21 (LVFFA): Highly hydrophobic, β-sheet prone
This region drives aggregation into amyloid fibrils
Mutations in this region (e.g., F19S) reduce aggregation
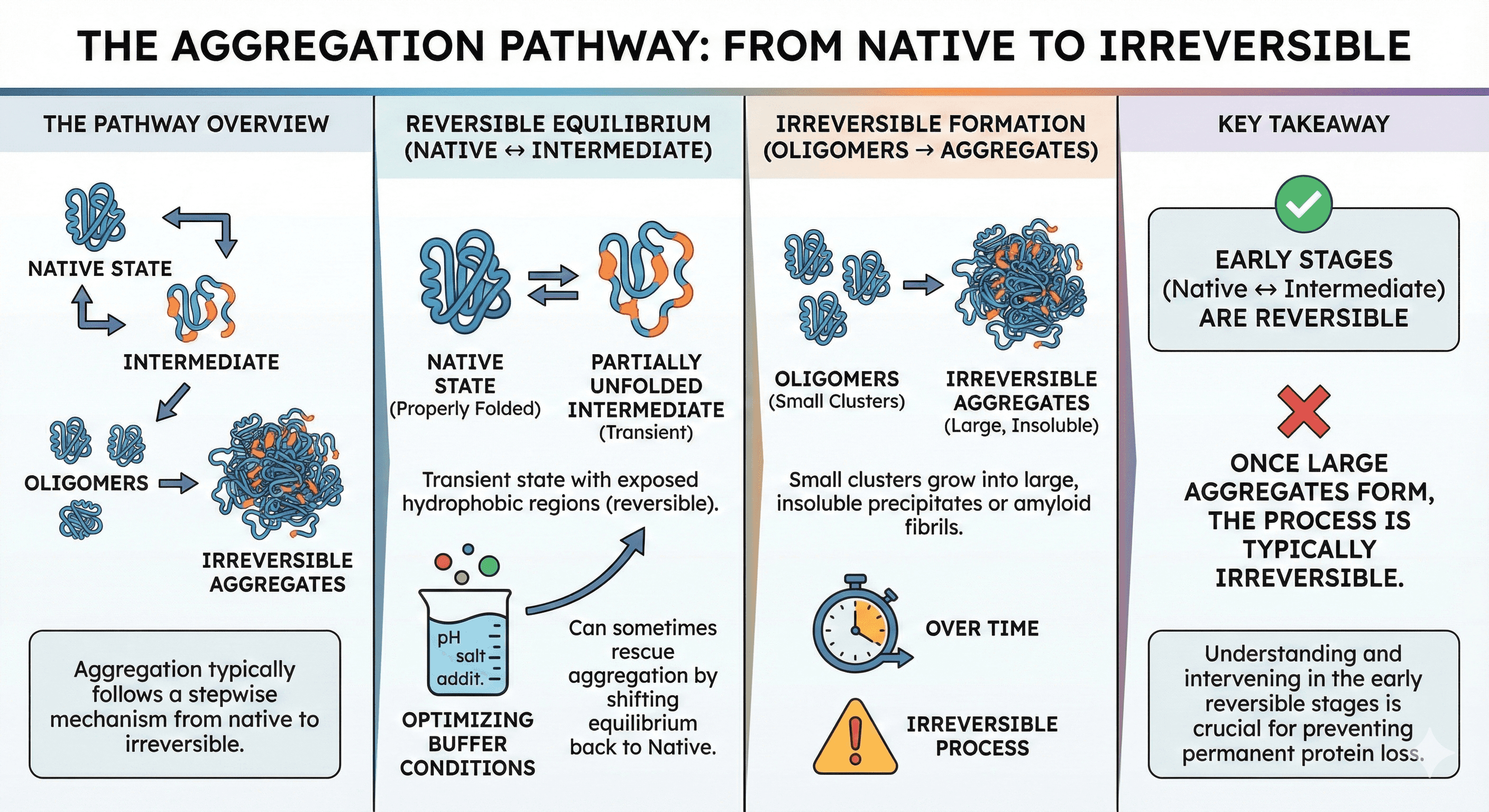
The Aggregation Pathway
Aggregation typically follows a stepwise mechanism:
Native ↔ Intermediate → Irreversible Aggregate
Native state: Properly folded, functional protein
Partially unfolded intermediate: Transient state with exposed hydrophobic regions (reversible)
Oligomers: Small clusters (dimers, trimers) that can grow
Irreversible aggregates: Large, insoluble precipitates or amyloid fibrils
The key is that early stages (native ↔ intermediate) are reversible—this is why optimizing buffer conditions can sometimes rescue aggregation. Once large aggregates form, the process is typically irreversible.
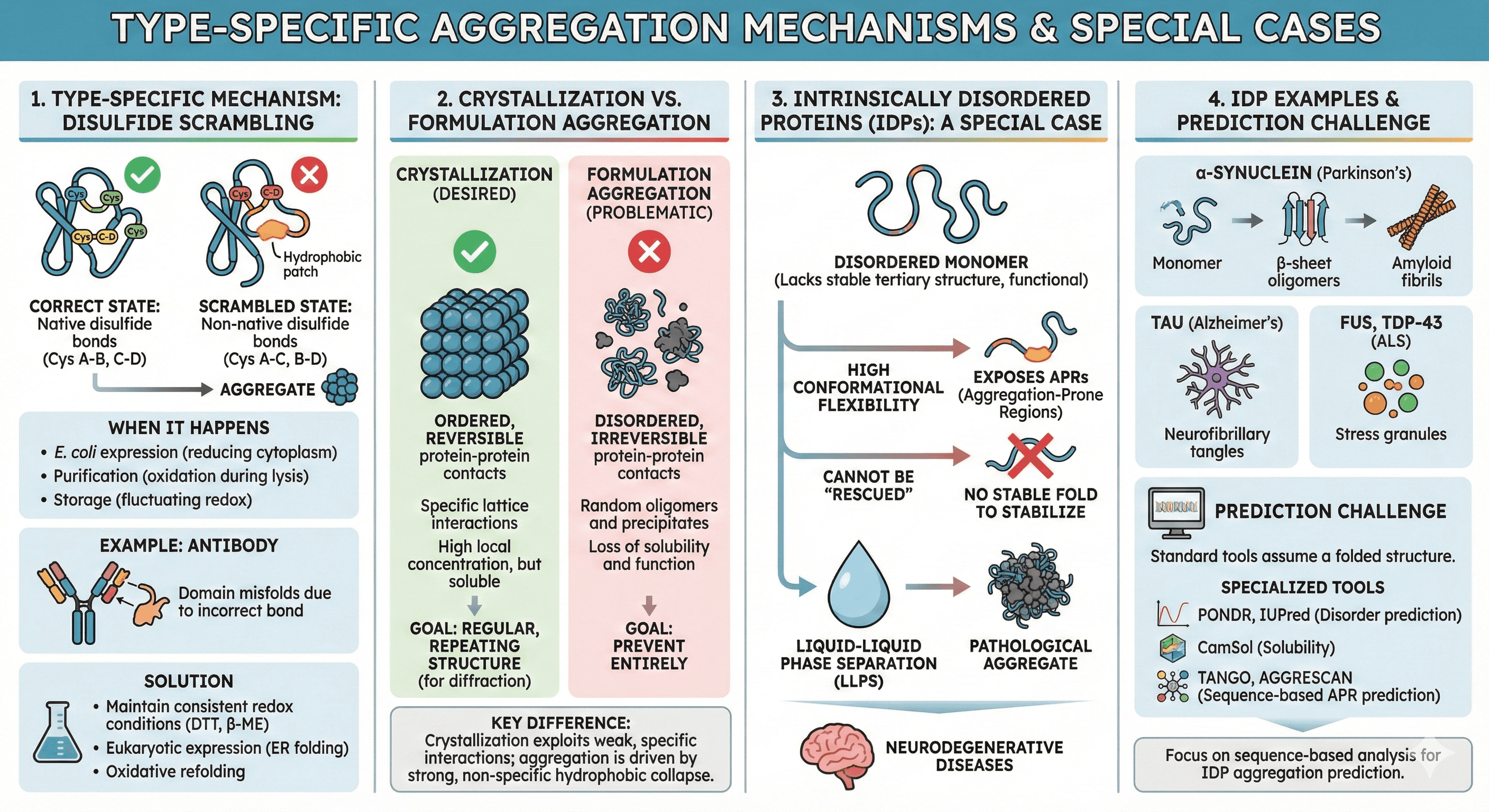
Type-Specific Aggregation Mechanisms
Disulfide scrambling (incorrect redox states): A major but often overlooked aggregation mechanism, especially for proteins with multiple cysteines:
Correct state: Native disulfide bonds (Cys A-Cys B, Cys C-Cys D)
Scrambled state: Non-native disulfide bonds (Cys A-Cys C, Cys B-Cys D)
Result: Misfolded protein with exposed hydrophobic patches → aggregation
When it happens:
During expression in E. coli (reducing cytoplasm, incorrect oxidation in periplasm)
During purification (oxidation during lysis, improper reducing conditions)
Storage with fluctuating redox conditions
Example: Antibody aggregation
IgG has 16 disulfide bonds
If even one forms incorrectly, the domain misfolds
Aggregates form through exposed hydrophobic regions
Solution:
Maintain consistent redox conditions (add DTT or β-mercaptoethanol)
For secreted proteins: Express in systems with proper oxidative folding (ER of eukaryotic cells)
Use oxidative refolding protocols if expressed in E. coli
Crystallization vs formulation aggregation:
These are distinct phenomena often confused:
Crystallization (desired):
Ordered, reversible protein-protein contacts
Specific lattice interactions
High local concentration in crystal, but soluble in mother liquor
Goal: Regular, repeating structure for diffraction
Formulation aggregation (problematic):
Disordered, irreversible protein-protein contacts
Random oligomers and precipitates
Loss of solubility and function
Goal: Prevent entirely
Key difference: Crystallization exploits weak, specific interactions; aggregation is driven by strong, non-specific hydrophobic collapse.
Intrinsically Disordered Proteins (IDPs): A Special Case
IDPs lack stable tertiary structure but are functional. They present unique aggregation challenges:
Why IDPs aggregate:
High conformational flexibility exposes aggregation-prone regions
Cannot be "rescued" by stabilizing mutations (no stable fold to stabilize)
Often phase-separate into droplets (liquid-liquid phase separation, LLPS) which can mature into aggregates
Examples:
α-synuclein (Parkinson's disease): Disordered monomer → β-sheet oligomers → amyloid fibrils
Tau (Alzheimer's): Disordered in solution, aggregates into neurofibrillary tangles
FUS, TDP-43 (ALS): Phase-separate into stress granules, can aggregate pathologically
Prediction challenge: Standard tools assume a folded structure. For IDPs, use specialized tools:
PONDR, IUPred (disorder prediction)
CamSol (solubility)
Focus on sequence-based APR prediction (TANGO, AGGRESCAN)
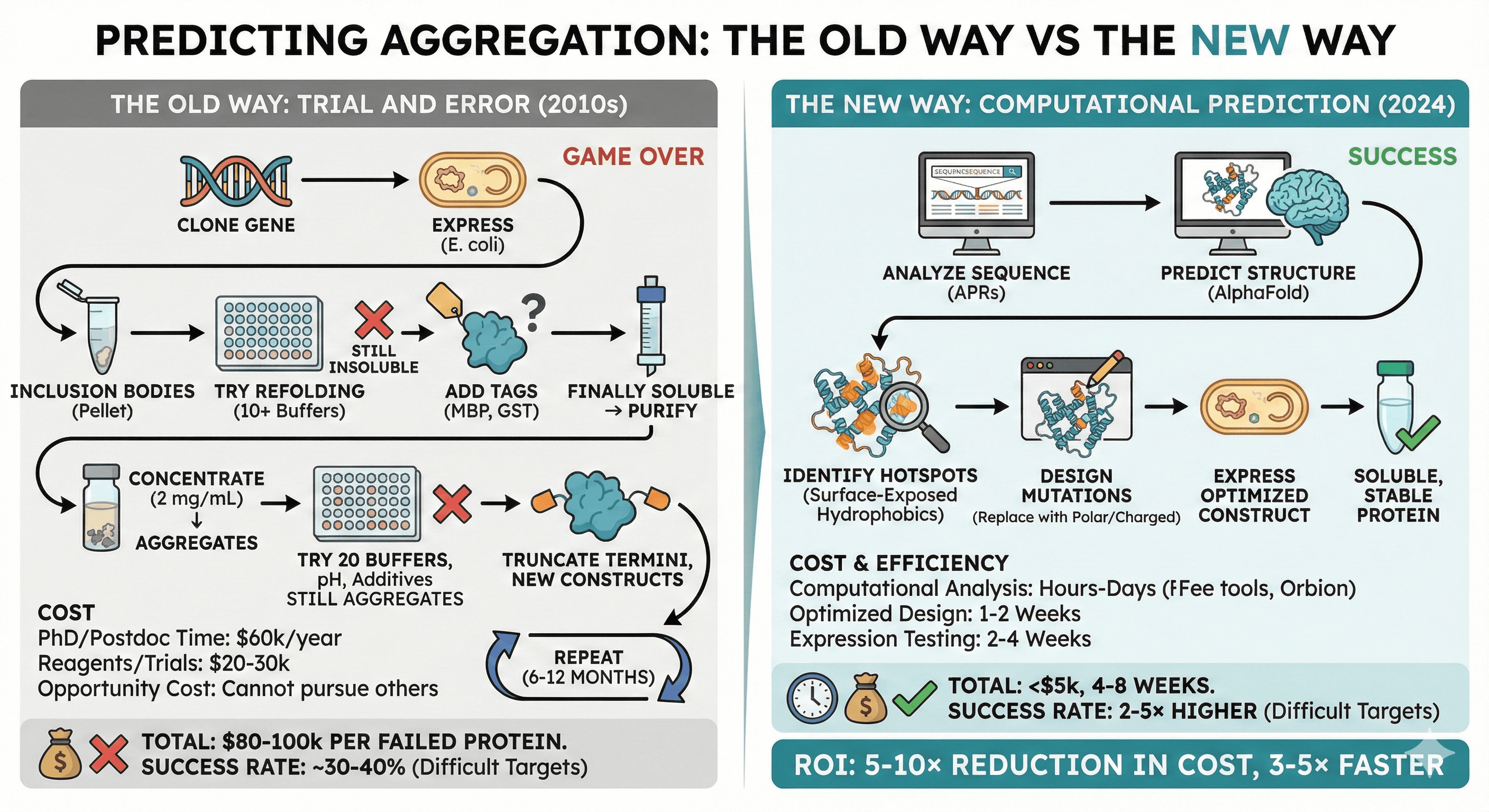
Predicting Aggregation: The Old Way vs The New Way
The Old Way: Trial and Error
Typical workflow (2010s):
Clone gene → Express in E. coli
Inclusion bodies → Try refolding (10+ buffer conditions)
Still insoluble → Try solubility tags (MBP, GST)
Finally soluble → Purify
Concentrate → Aggregates at 2 mg/mL
Try 20 buffer conditions, additives, pH values
Still aggregates → Truncate termini, try new constructs
Repeat for 6-12 months
Cost:
PhD student/postdoc time: $60k/year
Reagents, expression trials: $20-30k
Opportunity cost: Cannot pursue other targets
Total: $80-100k per failed protein
Success rate: ~30-40% for difficult targets (GPCRs, membrane proteins, intrinsically disordered proteins)
The New Way: Computational Prediction
Modern workflow (2024):
Analyze sequence for aggregation-prone regions (APRs)
Predict structure (AlphaFold if no experimental structure)
Identify hotspots: Surface-exposed hydrophobic patches
Design mutations: Replace aggregation-prone residues with polar/charged residues
Express optimized construct → Soluble, stable protein
Cost:
Computational analysis: Hours (free tools) to days (Orbion)
Optimized construct design: 1-2 weeks
Expression testing: 2-4 weeks
Total: <$5k, 4-8 weeks
Success rate improvement: 2-5× higher success rate for difficult targets (when computational design is used)
ROI: 5-10× reduction in cost, 3-5× faster
Tools for Aggregation Prediction
Sequence-Based Tools (No Structure Required)
1. AGGRESCAN
What it does: Scans sequence for aggregation-prone regions
Output: Aggregation propensity score per residue
Best for: Quick initial screen
Limitation: Doesn't account for burial in folded structure
2. TANGO
What it does: Predicts β-sheet aggregation (amyloid-like)
Output: Aggregation rate constants
Best for: Identifying amyloidogenic segments
Limitation: Focused on β-sheet aggregation (misses other types)
3. Zyggregator
What it does: Kinetic model of aggregation based on physicochemical properties
Output: Aggregation rate at different concentrations/temperatures
Best for: Predicting concentration dependence
Limitation: Empirical model, less accurate for membrane proteins
Structure-Based Tools (Requires 3D Structure)
4. AGGRESCAN3D
What it does: Maps aggregation propensity onto 3D structure
Output: Surface-exposed aggregation-prone patches
Best for: Identifying regions to mutate
Limitation: Requires accurate structure
5. CamSol (Cambridge Solubility Predictor)
What it does: Predicts protein solubility from structure
Output: Solubility score, identifies problematic residues
Best for: Antibody and therapeutic protein optimization
Limitation: Trained mostly on antibodies
6. Spatial Aggregation Propensity (SAP)
What it does: Analyzes clustering of hydrophobic residues on surface
Output: Hotspot map
Best for: Detecting non-obvious aggregation mechanisms
AI/ML Platforms (Structure + Sequence + Context)
7. Orbion
What it does:
Predicts aggregation hotspots from AlphaFold or experimental structure
Suggests specific mutations to reduce aggregation (with ΔΔG predictions)
Integrates with stability predictions (ensure mutations don't destabilize fold)
Recommends expression system based on aggregation risk
Output:
Aggregation risk score
Residue-level hotspot map
Mutation recommendations (e.g., "L47S reduces aggregation 80%, ΔΔG = +0.3 kcal/mol, maintains stability")
Best for: Rescue strategies for failed targets, therapeutic antibody optimization
Advantage: Combines multiple prediction methods + experimental validation data
How to Fix Aggregation: The Rescue Toolkit
Once you know why your protein aggregates, you can fix it. Here are the five main strategies:
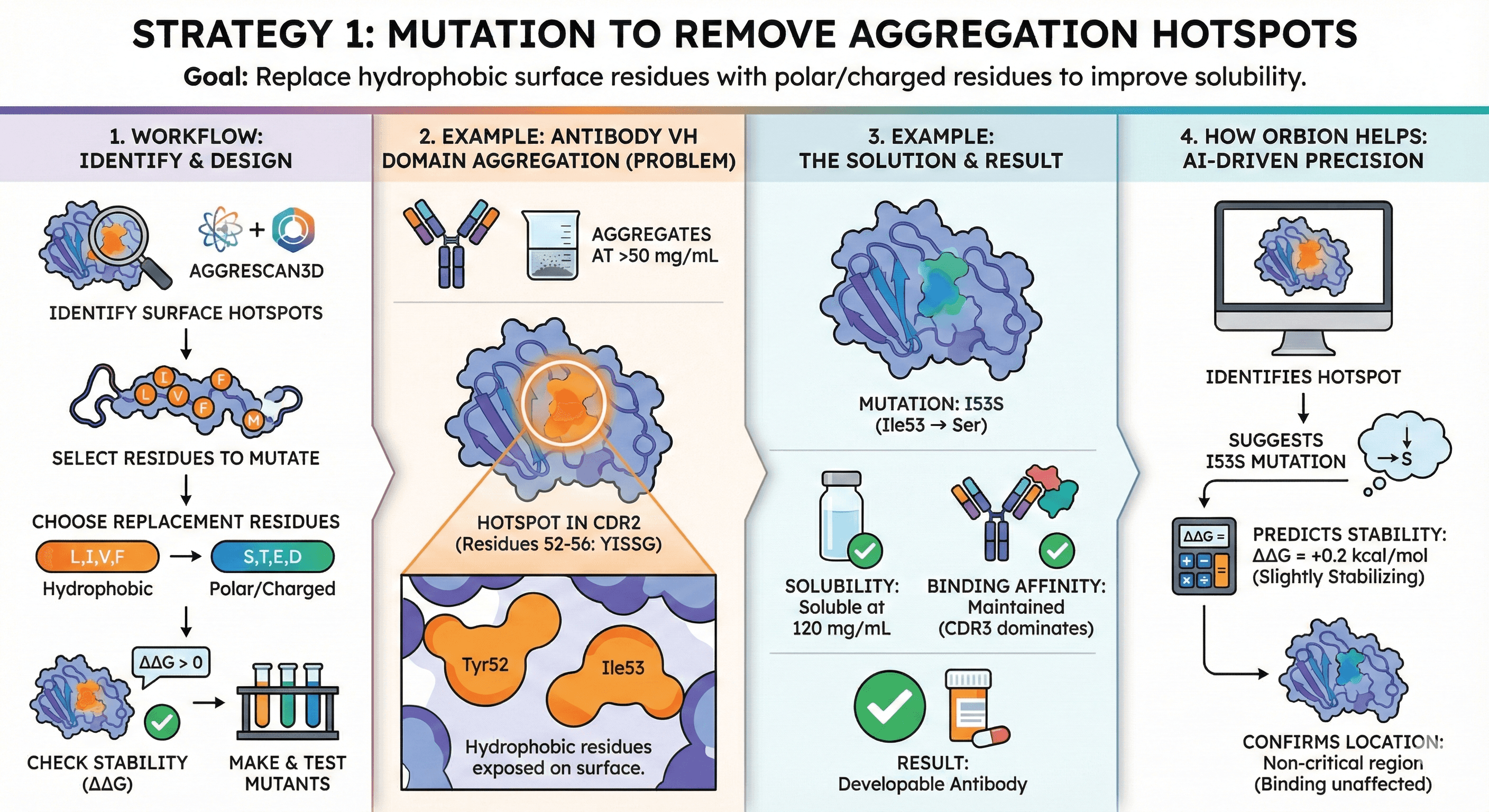
Strategy 1: Mutation to Remove Aggregation Hotspots
Goal: Replace hydrophobic surface residues with polar/charged residues
Workflow:
Identify surface-exposed hydrophobic patches (AGGRESCAN3D, Orbion)
Select residues to mutate (prioritize Leu, Ile, Val, Phe, Met on surface)
Choose replacement residues:
Hydrophobic → Polar: Leu → Ser, Ile → Thr
Hydrophobic → Charged: Val → Glu, Phe → Asp (if structure allows)
Check that mutation doesn't destabilize fold (Orbion ΔΔG prediction, or Rosetta)
Make 2-3 mutants, test in parallel
Example: Antibody VH Domain Aggregation
Problem:
Therapeutic antibody aggregates above 50 mg/mL
AGGRESCAN3D identifies hotspot in CDR2 (VH residues 52-56: YISSG)
Hydrophobic Tyr52 and Ile53 exposed on surface
Solution:
Mutate Ile53 → Ser (I53S)
Test: I53S variant remains soluble at 120 mg/mL
Binding affinity: Maintained (CDR3 dominates binding)
Result: Developable antibody
How Orbion helps:
Identifies the hotspot
Suggests I53S mutation
Predicts ΔΔG = +0.2 kcal/mol (slightly stabilizing)
Confirms mutation is in non-critical region (binding affinity unaffected)
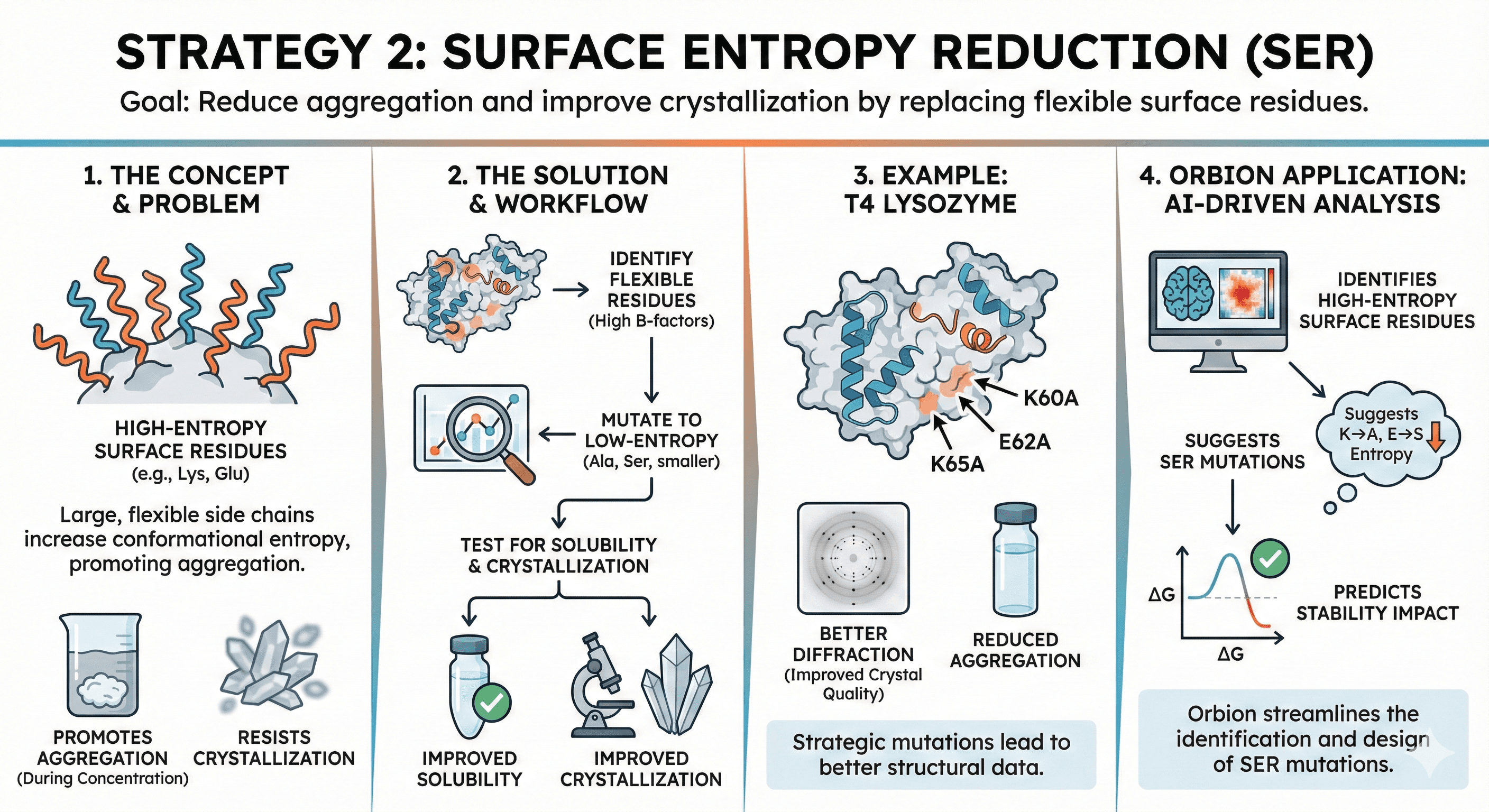
Strategy 2: Surface Entropy Reduction (SER)
Concept: High-entropy surface residues (Lys, Glu with flexible side chains) can promote aggregation by increasing conformational entropy. Replacing them with low-entropy residues (Ala, Ser) reduces aggregation and improves crystallization.
When to use: Proteins that aggregate during concentration or resist crystallization
How it works:
Identify surface Lys or Glu residues with high B-factors (flexible)
Mutate to Ala or Ser (smaller, less flexible)
Test for solubility and crystallization
Example: T4 Lysozyme
Mutation: K60A, E62A, K65A
Result: Improved crystal quality (better diffraction), reduced aggregation
Orbion:
Identifies high-entropy surface residues
Suggests SER mutations
Predicts impact on stability
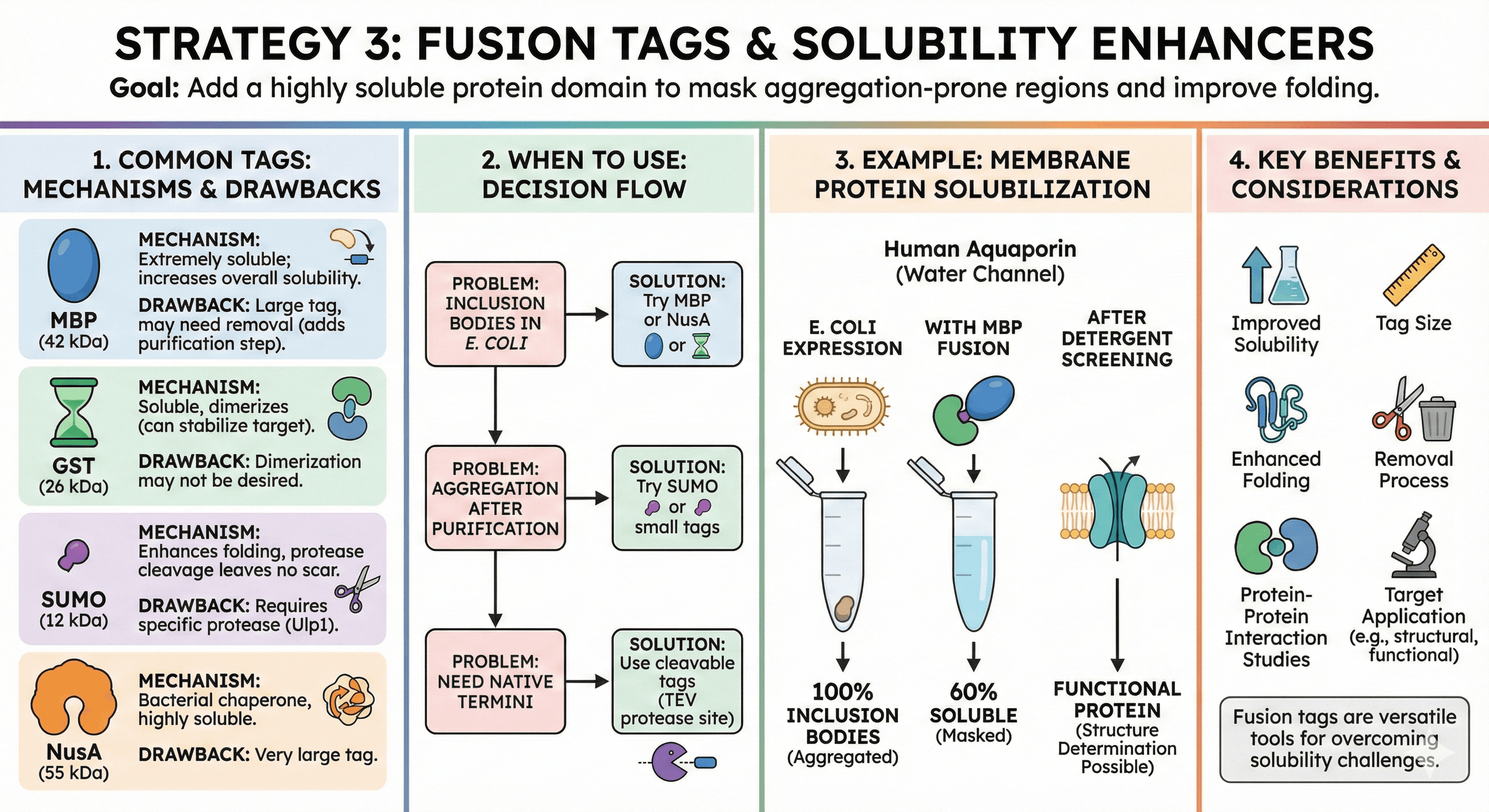
Strategy 3: Fusion Tags & Solubility Enhancers
Goal: Add a highly soluble protein domain to mask aggregation-prone regions
Common tags:
MBP (Maltose Binding Protein) - 42 kDa
Mechanism: MBP is extremely soluble; fusing it to your protein increases overall solubility
Best for: Cytoplasmic proteins, difficult-to-express proteins
Drawback: Large tag, may need removal (adds purification step)
GST (Glutathione S-Transferase) - 26 kDa
Mechanism: Soluble, dimerizes (can help stabilize target protein)
Best for: Protein-protein interaction studies, pull-down assays
Drawback: Dimerization may not be desired
SUMO (Small Ubiquitin-like Modifier) - 12 kDa
Mechanism: Enhances folding, protease cleavage leaves no scar
Best for: Proteins requiring native N-terminus
Drawback: Requires specific protease (Ulp1)
NusA - 55 kDa
Mechanism: Bacterial chaperone, highly soluble
Best for: Proteins prone to inclusion bodies
Drawback: Very large tag
When to use:
Inclusion bodies in E. coli → Try MBP or NusA
Aggregation after purification → Try SUMO or small tags
Need native termini → Use cleavable tags (TEV protease site)
Example: Membrane Protein Solubilization Human aquaporin (water channel):
In E. coli: 100% inclusion bodies
With MBP fusion: 60% soluble
After detergent screening: Functional protein
Result: Structure determination possible
Strategy 4: Expression System Optimization
Principle: Different organisms have different folding machinery, chaperones, and PTM capabilities. Switching systems can solve aggregation.
Decision tree:
E. coli (bacteria):
Pros: Fast, cheap, high yield
Cons: No eukaryotic chaperones, no glycosylation, limited disulfide formation
Best for: Simple cytoplasmic proteins, no PTMs needed
Pichia pastoris (yeast):
Pros: Eukaryotic folding, some glycosylation, disulfide bonds
Cons: High-mannose glycans (non-human), lower yield than E. coli
Best for: Secreted proteins, simple glycoproteins
Sf9/Hi5 (insect cells):
Pros: Good folding, complex disulfides, some glycosylation
Cons: Minimal sialylation, expensive
Best for: GPCRs, complex membrane proteins
HEK293/CHO (mammalian cells):
Pros: Native-like folding, full glycosylation, correct PTMs
Cons: Slow, expensive, lower yield
Best for: Therapeutic antibodies, membrane proteins requiring native PTMs
Example: GPCR Expression β2-Adrenergic receptor:
E. coli: Inclusion bodies (100%)
Pichia: Partially functional (20% yield)
Sf9 (insect cells): Functional receptor (60% yield)
Choice: Insect cells (balance of yield and function)
How Orbion helps:
Analyzes PTM requirements (glycosylation, phosphorylation)
Recommends expression system
Example: "This protein requires N-glycosylation at Asn120 → Use insect or mammalian cells"
Strategy 5: Buffer & Formulation Optimization
Goal: Optimize solution conditions to prevent aggregation
Key variables:
1. pH
Principle: Proteins aggregate near their pI (isoelectric point) due to reduced charge repulsion
Solution: Work at pH 1-2 units away from pI
Tool: Calculate pI (ExPASy ProtParam), test pH 5.5, 6.5, 7.5, 8.5
2. Salt concentration
Principle: Moderate salt (50-150 mM NaCl) screens charge interactions, reduces aggregation
Too low salt: Charge-charge aggregation
Too high salt: Salting-out effect (protein precipitates)
Optimal: 100-150 mM NaCl for most proteins
3. Additives
Arginine (50-500 mM): Suppresses aggregation (mechanism unclear, likely disrupts hydrophobic interactions)
Glycerol (5-20%): Stabilizes native state, reduces unfolding
Sucrose/Trehalose (5-10%): Osmolytes, protect against freeze-thaw
Detergents (membrane proteins): Maintain solubility (DDM, LMNG, amphipols)
4. Temperature
Cold storage (4°C): Slows aggregation kinetics
Flash-freezing: Rapid cooling minimizes ice crystal damage
Avoid freeze-thaw: Each cycle causes partial unfolding
Example: Antibody Formulation Optimization Monoclonal antibody aggregating at >100 mg/mL:
Initial buffer: PBS (pH 7.4, 150 mM NaCl)
Aggregates at 120 mg/mL
Optimization screen:
pH: Test 5.5, 6.5, 7.5, 8.5 → Best at pH 6.0
Additives: Add 10 mM arginine + 5% sucrose
Result: Stable at 150 mg/mL for 6 months
Case Study: Rescuing an Aggregation-Prone Therapeutic Antibody
The Challenge
Target: Therapeutic monoclonal antibody (IgG1) for cancer Problem: Aggregates above 50 mg/mL (subcutaneous formulation requires >100 mg/mL) Timeline: 8 months in, Phase I trials delayed
The Investigation
Step 1: Identify aggregation mechanism
DLS (dynamic light scattering): Oligomers form at >30 mg/mL
SEC-MALS: High-molecular-weight species (dimers, trimers)
Conclusion: Concentration-dependent, reversible aggregation
Step 2: Computational prediction
Run AGGRESCAN3D on antibody structure
Hotspot identified: VH CDR2 (residues 52-56)
Hydrophobic patch (Tyr52, Ile53, Phe55) exposed on surface
Step 3: Rational mutagenesis
Design mutations:
I53S (Ile → Ser)
F55T (Phe → Thr)
Double mutant: I53S/F55T
Predict ΔΔG (stability): Orbion predicts +0.3 kcal/mol (slightly stabilizing)
The Solution
Step 4: Experimental validation
Express I53S, F55T, and I53S/F55T variants
Test solubility at 100, 150, 200 mg/mL
Results:
Wild-type: Aggregates at 60 mg/mL
I53S: Soluble to 130 mg/mL
F55T: Soluble to 110 mg/mL
I53S/F55T: Soluble to 180 mg/mL ✓
Step 5: Functional validation
Binding affinity (SPR): I53S/F55T binds with same Kd as wild-type
ADCC activity: Maintained
Pharmacokinetics (mouse model): Half-life unchanged
Outcome:
Developable antibody formulation (150 mg/mL)
Phase I trials resumed
Time saved: 12 months (avoided complete redesign)
The Economics of Aggregation Prevention
Cost of Failure
Academic target:
1 year of postdoc time: $60k
Reagents: $20k
Opportunity cost: Missed publications, delayed graduation
Total: $80k
Therapeutic antibody:
Discovery program: $10-20M
If aggregation detected late (Phase I): $50-100M wasted
Delay to market: 1-2 years = $500M-1B in lost revenue
ROI of Computational Prediction
Upfront investment:
Orbion subscription: $X/month
Computational time: 1-2 days
Savings:
Avoid 6-12 months of trial-and-error
Test 2-3 designed mutants instead of 50 random constructs
Increase success rate from 30% → 70%
ROI: 10-20× return on investment
Practical Checklist: Preventing Aggregation
Before Expression
[ ] Run aggregation prediction (AGGRESCAN, Orbion)
[ ] Check pI (avoid working near pI)
[ ] Identify surface hydrophobic patches
[ ] Design 2-3 mutants if hotspots found
[ ] Choose expression system based on PTM needs
During Expression
[ ] Small-scale test (don't commit to 10L culture immediately)
[ ] Check soluble vs insoluble fractions (SDS-PAGE)
[ ] If insoluble: Try 16°C expression, co-express chaperones, or add fusion tag
[ ] If soluble but low yield: Optimize induction conditions (IPTG concentration, time)
During Purification
[ ] Keep protein cold (4°C throughout)
[ ] Add stabilizers to buffer (glycerol 10%, DTT if cysteines present)
[ ] Avoid high protein concentrations early (dilute after elution)
[ ] Run SEC (size-exclusion chromatography) to remove aggregates
During Storage
[ ] Aliquot and flash-freeze (minimize freeze-thaw cycles)
[ ] Add cryoprotectants (10% glycerol or 5% trehalose)
[ ] Store at -80°C (more stable than -20°C)
[ ] For long-term: Lyophilize (freeze-dry)
If It Still Aggregates
[ ] Try fusion tag (MBP, SUMO)
[ ] Switch expression system (E. coli → yeast → insect → mammalian)
[ ] Revisit construct design (truncate disordered regions)
[ ] Screen formulation buffers (pH, salt, additives)
The Future: AI-Driven Aggregation Rescue
Machine learning models trained on millions of protein sequences and structures are making aggregation prediction more accurate.
Emerging Tools
1. AlphaFold confidence scores as aggregation predictors
Low pLDDT regions (<70) often correlate with aggregation-prone loops
Not explicit aggregation prediction, but useful heuristic
2. Generative models for solubility optimization
Tools like ProteinMPNN can redesign surface to maximize solubility
Generate variants with same function, improved expression
3. Deep learning scoring functions
Train on experimental aggregation data
Predict aggregation rate at different concentrations
4. Integration platforms (like Orbion)
Combine structure, sequence, PTM prediction, stability
Suggest complete rescue strategy (mutations + expression system + formulation)
The Bottom Line
Protein aggregation is not random bad luck. It is a predictable, solvable engineering problem.
The old paradigm: Express → Hope it works → Troubleshoot for months
The new paradigm: Predict → Design → Express optimized construct → Success
With modern computational tools, you can:
Predict aggregation before expressing anything
Design rescue mutations to remove hotspots
Choose the right expression system based on PTM and folding needs
Optimize formulation computationally before experimental screening
The payoff:
2-5× improvement in success rates
3-5× faster timelines
10× cost reduction
The difference between a "difficult protein" and a "successful structure" is often just 1-2 well-placed mutations identified by prediction tools like Orbion.
Aggregation used to be the graveyard of protein projects. Today, it's dramatically reduced—if you use the right tools. While not every aggregation problem can be solved computationally, the success rate has improved dramatically, transforming once-intractable targets into achievable goals.
Ready to Rescue Your Aggregating Protein?
If you have a protein that aggregates during expression, purification, or storage, Orbion can help identify why and suggest how to fix it.
Orbion provides:
Aggregation hotspot prediction from structure or sequence
Specific mutation recommendations (with ΔΔG stability predictions)
Expression system selection based on PTM requirements
Formulation optimization guidance
Book a 20-Minute Demo
Sign up free for unlimited Overview runs — summary, sequence-based analysis, homology search. For the full Characterization — PTMs, binding sites, stability variants, construct design — book a demo and we'll run your target live.
