Blog
Orbion Team
The 6 Cases Where pLDDT Confidence Misleads You

You opened your AlphaFold model in PyMOL, saw a deep-blue α-helix at pLDDT 92, and used it to define a truncation boundary for crystallization—or worse, to pick the mutational hotspot for your next library. Six months and a dozen failed constructs later, biophysics tells you the "helix" is unstructured in solution, the "domain" only folds when bound to a partner you didn't model, and the engineered chimera you designed around that confident region falls off the bench in inclusion bodies. The pLDDT score wasn't lying. You were asking it the wrong question.
pLDDT is a per-residue local-distance accuracy estimate, nothing more. Treating it as a proxy for biological reality is one of the most expensive habits in computational structural biology today. Below are the six failure modes that most reliably catch even careful researchers off guard, how to recognize them before you commit resources, and the orthogonal evidence that distinguishes a confidently-correct prediction from a confidently-wrong one.
Key Takeaways
Shallow-MSA proteins inherit common-fold confidence: pLDDT can reach >85 on regions where AlphaFold has effectively guessed the family fold rather than learned your sequence's structure.
Disorder-to-order transitions hide behind high pLDDT: motifs that fold only upon binding (MoRFs, SLiMs, fly-casting helices) routinely score 70–90 in the apo prediction.
Multi-state proteins collapse to a single high-confidence state: domain-swapped dimers, fold-switchers, and conformational ensembles are forced into one model with no flag that alternatives exist.
Membrane protein topology errors coexist with confident TM helices: pLDDT scores the helix as well-folded while the inside/outside orientation may be reversed or jumbled.
Engineered chimeras and circular permutants get component-level confidence: AlphaFold has seen each donor domain in training, so each piece scores high—but the interface between them is invented.

What pLDDT Actually Measures (And Doesn't)
Before diagnosing failure modes, it's worth being precise about what the score represents. pLDDT is a per-residue prediction of the local Distance Difference Test (lDDT) score (Mariani et al., 2013), defined as the fraction of inter-atomic distances within a 15 Å radius of each Cα that are preserved within four tolerance thresholds (0.5, 1, 2, and 4 Å) compared to the true structure. AlphaFold's network predicts this score because lDDT is a local metric—it does not penalize global rigid-body errors—and so pLDDT inherits exactly that property.
This has three immediate consequences that researchers routinely forget:
pLDDT is local, not global. A residue at pLDDT 95 sits in a locally consistent environment. It says nothing about whether the entire domain is in the right rotation or translation relative to the rest of the chain. That is PAE's job.
pLDDT is structural, not biological. The score has no concept of cofactors, partners, membranes, or solution conditions. It reflects what the network believes is locally self-consistent given the inputs it actually saw.
pLDDT is averaged across the recycled outputs. Because AF2 recycles the structure module multiple times, pLDDT reflects convergence of the network on a self-consistent solution, not the agreement of that solution with experimental reality.
The score is therefore best understood as a self-consistency confidence, not a correctness confidence. Most of the time these coincide. The six cases below are the cases where they don't.
Why "High pLDDT but Wrong" Is Common, Not Rare
pLDDT was designed to predict the local Distance Difference Test score (Mariani et al., 2013)—the agreement of inter-atomic distances within 15 Å of each Cα. It is a measure of how well the network has converged on a self-consistent local geometry, not a measure of how well that geometry matches biological truth.
There are at least three structural reasons high pLDDT does not equal correctness:
Convergent fold memorization. When the MSA suggests a fold AlphaFold has seen many times, the network can produce a locally consistent structure even with weak evidence from the target sequence itself. Self-consistency is high; biological fidelity may not be (Buel & Walters, 2022).
Apo-only predictions. AF2 predicts the protein alone—no ligand, no metal, no PTM, no partner. Conformations that exist only in the holo state cannot be scored against the apo prediction's confidence (Saldaño et al., 2022).
Single-state output. A single best model is emitted (or five recycled variants of essentially the same minimum), so multi-state systems lose their alternative conformations without any score reporting that loss (Akdel et al., 2022).
The six cases that follow are the practical consequences of these three structural limitations.
Case 1 — Shallow-MSA Proteins Inheriting Common-Fold Confidence
What goes wrong
When AlphaFold is given a sequence with a thin MSA (≤30 effective sequences, Neff/L < 0.5, or a sequence drawn from an underrepresented organism or de novo-designed scaffold), the evoformer has little co-evolutionary signal to work with. Yet the structure module is still asked to emit a model. What you frequently get is a generic family fold—a Rossmann, a TIM-barrel, an immunoglobulin sandwich, a four-helix bundle—rendered with surprisingly high pLDDT. The network has memorized that fold from training, and in the absence of contradicting evidence it defaults to it.
This is most insidious for proteins that are real but rare: orphan ORFs from non-model genomes, ancient enzymes from extremophiles, designed binders, and viral proteins from emerging clades. The pLDDT can read 80–90 in the core while the actual fold may be entirely different—or, more commonly, the secondary structure topology is right but specific contacts and active-site geometry are mis-placed by 3–6 Å.
How to recognize the failure
Diagnostic | What you're looking for |
|---|---|
MSA depth (Neff/L) | Less than ~1 raises strong suspicion; less than 0.3 is dangerous |
Per-residue pLDDT and PAE | High pLDDT with patchy or asymmetric PAE within a single domain |
Multiple seeds / multiple ranks | If rank_0 through rank_4 give meaningfully different topologies, the prediction is unstable despite confidence |
Family ambiguity | If the top hit by HHsearch is < 30% identity to anything in PDB, the fold confidence is mostly prior, not posterior |
Disagreement with ESM-Fold or RoseTTAFold | Two models trained on different objectives diverging on the same input is a strong red flag |
What to trust, what not to
Trust | Don't trust |
|---|---|
The presence of secondary structure elements as approximate | Specific residue–residue contacts |
The general handedness and topology if multiple seeds agree | Active-site geometry, catalytic distances, exposed-vs-buried assignments |
Surface convex hull (for SAXS comparison) | Pocket shapes, druggability calls, mutation design at the active site |
The shallow-MSA case is also where Akdel et al.'s community assessment showed the largest gap between predicted and actual accuracy (Akdel et al., 2022): for sequences with effective MSA depth below ~30, real lDDT can lag pLDDT by 10–20 points. The score is reporting its self-belief, not its truth.
Remedy: Run subsampled-MSA ensembles, compare against ESM-Fold and an unbiased de novo predictor, and cross-check with SAXS or limited proteolysis before designing anything. If MSA depth is the root cause, an AstraUNFOLD review of disorder and topology often reveals that what AlphaFold called a confident loop is in fact a flexible region the network filled in with prior structure.
Concrete example. A de novo designed binder scaffold, built from a four-helix bundle motif and submitted without a partner, will routinely return pLDDT 88–92 across the bundle. The MSA contains only the designed sequence itself plus a handful of close homologs from the design family. The prediction is essentially a memorized scaffold rendering. When biophysics shows the binder is partially molten or oligomerizes off-target, the experimentalist is surprised—but the pLDDT was never reporting on the novel parts of the design (the binding face), only on the well-known scaffold backbone.
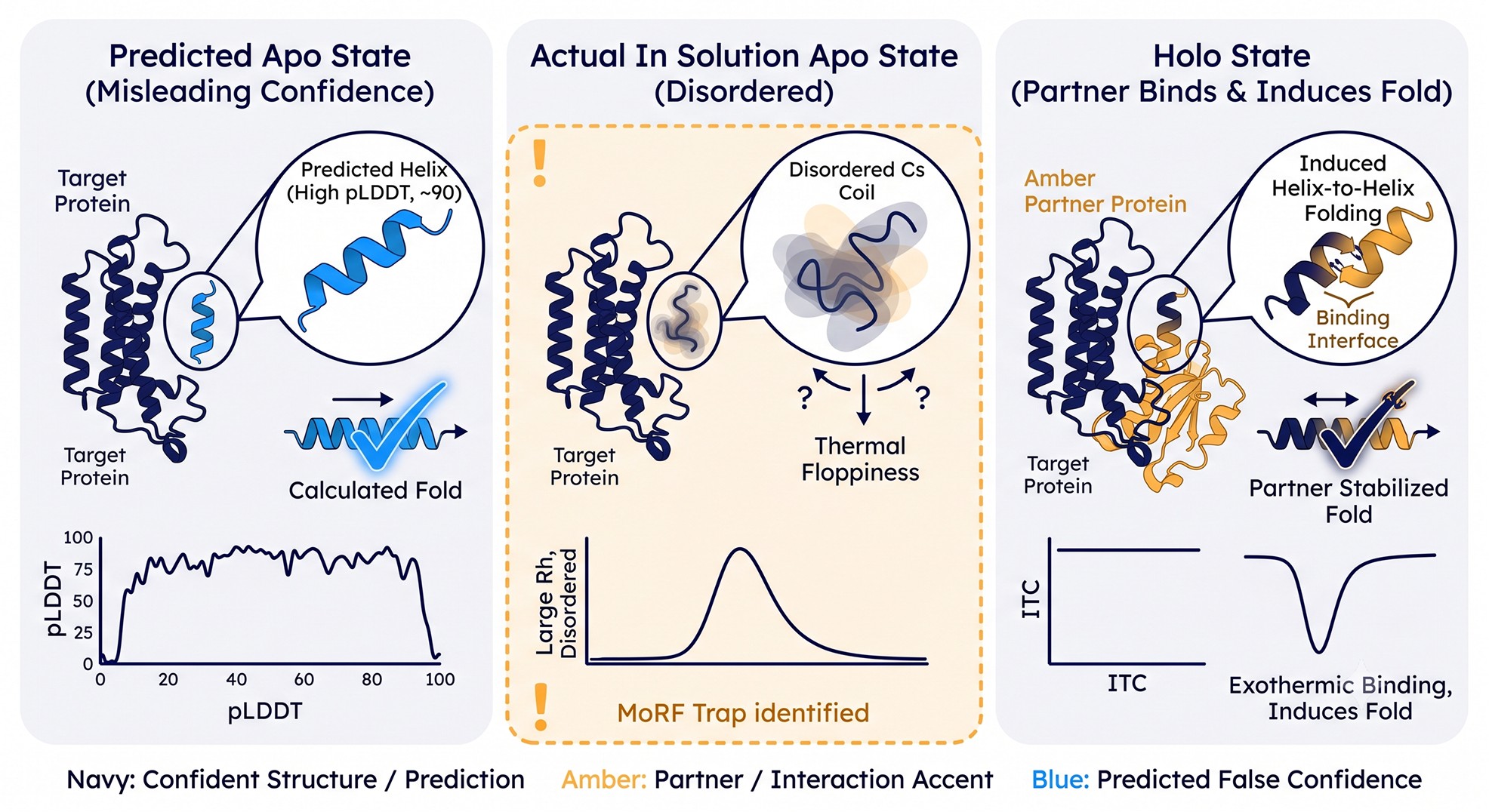
Case 2 — Disorder-to-Order Transitions (MoRFs, SLiMs, Fly-Casting Helices)
What goes wrong
A class of biologically critical motifs—Molecular Recognition Features (MoRFs), Short Linear Motifs (SLiMs), and induced-folding helices—are intrinsically disordered in solution but adopt defined structure only upon binding a partner. Classic examples: the C-terminal transactivation domain of p53 (folds on Mdm2 binding), the kinase-inducible domain (KID) of CREB (folds on KIX), and the disordered tails of many transcription factors that form short α-helices on coactivators.
When you predict the protein alone, AF2 frequently reports these motifs at pLDDT 70–88—not the <50 you'd expect for "true" disorder. The network has seen the bound conformation in the training set (the partner-bound complexes are in the PDB) and produces the folded form even though it's biologically irrelevant in isolation.
This is the trap that catches biophysicists most often. You see a confident helix in your prediction, you design a truncation that excludes the surrounding "noise," you express it—and the construct is unstructured by CD, smears by SEC, and aggregates within hours. The helix was real but conditional. You removed the partner that stabilized it.
How to recognize the failure
Sequence composition flag. Low hydrophobicity, high P/E/K/S content, and high charge fraction in a region predicted as helical is a giveaway. Run IUPred3 or PONDR-VSL2; if they say "disordered" but pLDDT says "folded," the motif is likely a MoRF.
PAE asymmetry. A short helix with high intra-helix pLDDT but high PAE to the rest of the chain is conformationally adrift—often the signature of a MoRF anchored only by its partner.
MoRFchibi or ANCHOR2 score. Dedicated MoRF predictors will flag these regions even when pLDDT is high.
AlphaFold-Multimer test. If you predict the protein with its known partner and the same region's confidence stays similar, it's truly folded. If confidence and contact geometry change significantly, the apo prediction was misleading.
Conservation pattern. SLiMs are short (3–12 residues), conserved at specific positions but with variable flanking residues. A confident helix that overlaps an annotated ELM motif is almost certainly a MoRF.
Solvent accessibility paradox. Many MoRFs are predicted as helices with hydrophobic faces exposed to solvent—because in reality those faces are buried against the partner. Exposed hydrophobic faces with high pLDDT is a near-certain MoRF signature.
What to trust, what not to
Treat any high-pLDDT region with disorder-prediction disagreement as conditionally folded. Do not truncate it without functional rationale. Do not assume the helical geometry you see is the geometry in solution. The AstraUNFOLD module in Orbion specifically flags MoRFs and conditional-fold regions by combining pLDDT with composition-based disorder and PAE coherence—precisely so that pLDDT alone never drives a truncation decision.
Why this happens mechanistically. The PDB is enriched for bound conformations of regulatory motifs because they were solved in complex with their partners. AlphaFold's training therefore associates these sequences with folded geometry. The network has no way of knowing that the partner is missing from your prediction input. The result is a confidently-folded apo structure for a sequence that, in solution alone, populates an extended ensemble. p53's TAD region predicted bound to MDM2 is the canonical example: in isolation, p53 1–93 is a textbook IDR; bound, it forms an amphipathic helix that AlphaFold reproduces faithfully.
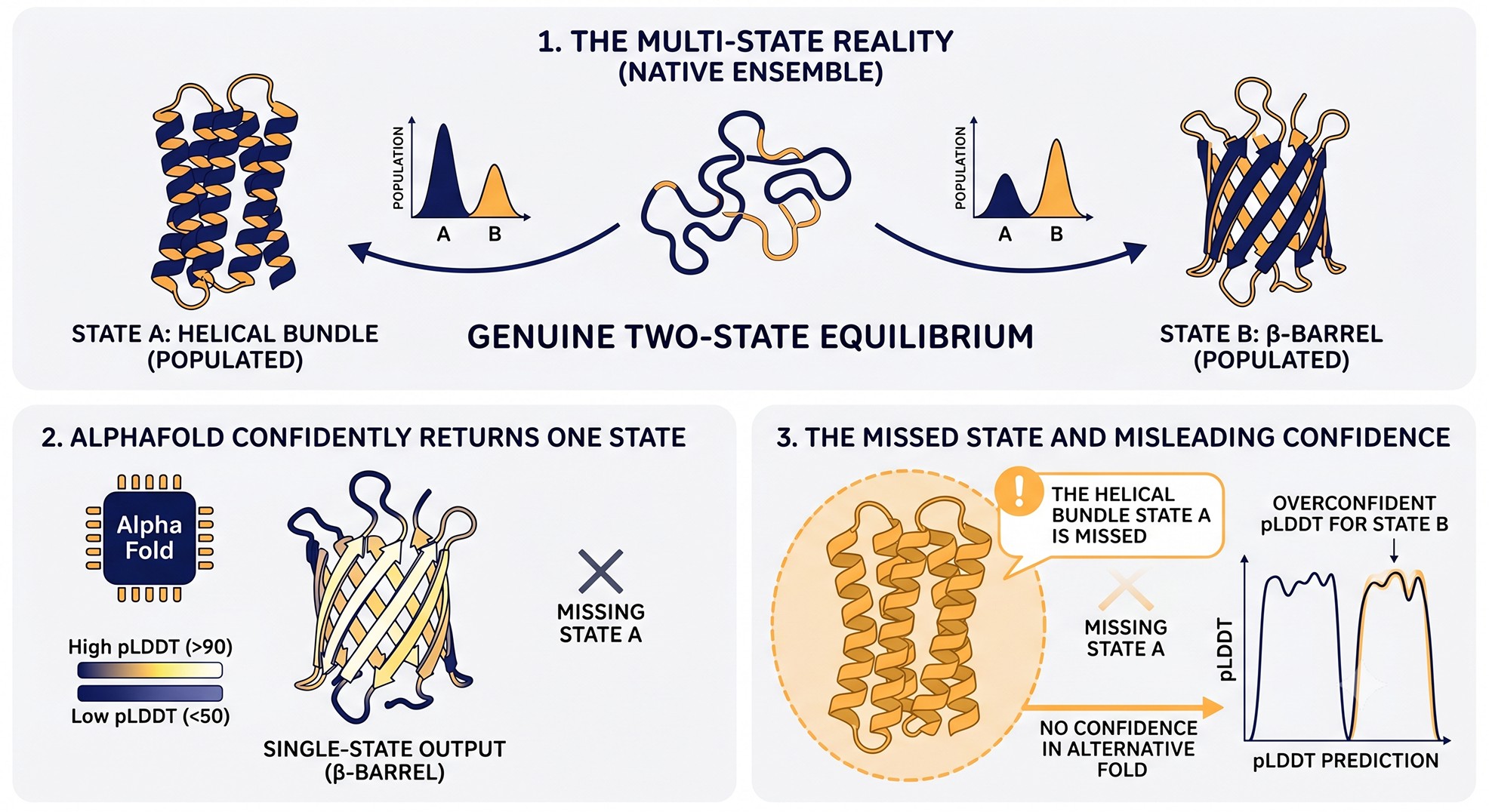
Case 3 — Domain Swapping, Fold-Switching, and Multi-State Proteins
What goes wrong
Some proteins exist in two or more genuinely distinct folds. RfaH switches between an α-helical bundle and a β-barrel depending on context (Burmann et al., 2012); KaiB exchanges between thioredoxin-like and dimeric folds; many chemokines and serpins undergo large conformational rearrangements; numerous oligomers form via domain swapping where the same monomer can fold into either a closed monomer or an open swapped dimer. AlphaFold's training objective collapses this multiplicity into a single output. Whichever conformation dominates the PDB wins, and pLDDT for that one state can be exquisite (>90).
The trap: you assume the prediction represents the structure. You design a mutation that stabilizes that conformation, only to discover the protein's function depended on the equilibrium between states. Or you design a construct that should crystallize the predicted closed monomer, and instead you get the swapped dimer at equilibrium—because that's actually the lower-energy state in your conditions.
How to recognize the failure
Diagnostic | What you're looking for |
|---|---|
Multiple ranks disagree on topology | rank_0 closed, rank_3 swapped; both at high confidence |
Subsampled MSAs | Reducing MSA depth often reveals alternative conformations as AF "loses" the dominant prior |
Known fold-switcher families | RfaH/NusG, lymphotactin, KaiB, serpins, MAD2, prion proteins |
Native MS or AUC heterogeneity | Multi-state oligomeric distribution suggests conformational exchange |
SEC peak doubling or shoulder | Often the empirical hallmark of swap/open–close equilibria |
What to trust, what not to
Trust the predicted fold as one valid state. Do not assume it is the only or dominant state. Test orthogonally with native MS, AUC, NMR HSQC (peak doubling is diagnostic), or HDX-MS. For known multi-state systems, run AF-cluster or MSA subsampling to surface alternative conformers; if AF2 produces two clearly different high-confidence models, that's evidence the protein is genuinely bistable, not that AF is unreliable.
The kinase example. Most kinases populate at least two functional states (DFG-in/active and DFG-out/inactive) and often a third (αC-out). AlphaFold typically predicts whichever state dominates the kinase's representation in the PDB—usually the inactive state, sometimes the active. Both states regularly come back with pLDDT > 90. Designing an ATP-competitive inhibitor against the predicted state is reasonable; designing a Type II inhibitor against a kinase whose DFG-out conformation was not predicted is a frequent and expensive mistake. The same logic applies to GPCRs (active vs inactive), nuclear receptors (apo vs agonist-bound vs antagonist-bound), and most allosteric enzymes.

Case 4 — Membrane Proteins with Confident Helices but Wrong Topology
What goes wrong
For membrane proteins, AF2 will reliably predict TM helices at very high pLDDT (often >90). What it does not score is whether each helix is correctly oriented relative to the bilayer—inside-out, outside-in, or in the right order. The model has no membrane in it. The structure module places helices into a bundle that is locally consistent, but the topology (N-in vs N-out, the number of TM passes, the position of re-entrant loops) is a separate determination that pLDDT does not capture.
Specific failure modes include:
Mis-numbered TM count. A 7-TM GPCR predicted with 6 or 8 confident helices, with the extra/missing one being a re-entrant loop or amphipathic helix.
Inverted N/C termini. N-in vs N-out flipped, which means your tagged construct has the tag on the wrong side of the membrane.
Confused re-entrant loops. Half-membrane-crossing segments (common in transporters and ion channels) confidently rendered as full TM helices, with the consequence that the substrate-binding pocket is in the wrong location.
Pseudo-symmetric mis-assignment. Internal pseudo-symmetric transporters (MFS, LeuT fold) predicted with the wrong rocker-switch state, even though every helix is high-pLDDT.
How to recognize the failure
Cross-check topology predictions. DeepTMHMM, TMHMM2.0, Phobius, or TOPCONS often disagree with AF on TM count and orientation. A disagreement is a flag.
Hydrophobicity profile. Kyte-Doolittle or biological hydrophobicity scales should align with TM segments. Where they don't, the AF-assigned helix may be cytoplasmic or extracellular.
PAE between TM helices. If PAE between helices is high (>15 Å) the bundle geometry is uncertain even if individual helices are confident.
Compare against the closest experimental structure. For GPCRs, transporters, and channels, structural homologs almost always exist. RMSD > 4 Å in the bundle is meaningful.
Check the polarity of solvent-exposed faces. Polar residues on lipid-facing surfaces is a topology error giveaway.
Glycosylation site placement. Annotated N-linked glycosylation sites should be on the extracellular side. If AF places them intracellularly, topology is inverted.
Positive-inside rule. The N-side of TM helices tends to be enriched in K/R residues (von Heijne's rule). Violation suggests topology error.
What to trust, what not to
Trust | Don't trust |
|---|---|
Existence and approximate length of TM helices | TM count if it conflicts with topology predictors |
Helix-helix packing for paralogs of solved structures | N/C terminus side of membrane |
Side-chain identity in the bundle interior | Substrate-pocket location for transporters |
Re-entrant loop assignments | |
Active state vs inactive state in GPCRs |
The PAE Insight Engine in Orbion is particularly useful here because it surfaces inter-helix PAE alongside the topology call from AstraUNFOLD, so the disagreement between "the helices are confident" and "the bundle's arrangement isn't" is visible at a glance.
Case 5 — Loops That Pack Artifactually into Surrounding Helices
What goes wrong
A common failure mode in moderate-MSA proteins: a long loop (15–40 residues), genuinely flexible or disordered in solution, gets packed by the structure module into the groove between two surrounding α-helices. The loop adopts a "stuffed-in" conformation that produces locally consistent geometry and high pLDDT (often 70–88), simply because the network found a low-energy local solution.
In reality the loop is dynamic. It samples many conformations, none of them stably packed. But because the predicted conformation is locally compatible with the rest of the structure, pLDDT does not flag the artifact. You then design a mutation in the loop to "stabilize" a contact you see—and discover by HDX-MS that the loop is fully solvent-exchanging in seconds, meaning your stabilizing mutation has no surface area to act on.
How to recognize the failure
Length and composition heuristic. Loops > 12 residues with > 30% charge or proline content are flexibility-prone.
Local PAE. A confidently-packed loop should show low PAE to the helices it contacts. If PAE within the loop is low but PAE to the helices is high (>10 Å), the packing is local-only and the overall position is unreliable.
B-factor analog. Cross-reference to crystal structures of the homolog: if the equivalent loop is high-B-factor or absent from electron density, AF's packed conformation is probably wrong.
HDX-MS or NMR R₂. Exchange rates and relaxation times tell you immediately whether a region is dynamic.
What to trust, what not to
Treat any long loop, no matter the pLDDT, as a candidate for flexibility. Trust the sequence position of the loop; treat the conformation as one possible state among many. Don't design point mutations in such loops to "lock in" contacts unless you have independent evidence the loop is structured.
Case 6 — Engineered Chimeras and Circular Permutants
What goes wrong
When you submit a chimera, a fusion, a circular permutant, or a heavily-engineered variant, AlphaFold processes the sequence against its training data and finds matches for the individual donor domains. Each domain folds confidently in isolation. The interface between donor components—the linker, the junction, the permuted termini—is not in the training set. The model fills it in with a plausible-looking but invented geometry, often at surprisingly high pLDDT.
This is where the AF2 prediction looks gorgeous and the cloned construct expresses as inclusion bodies. You designed the linker length based on the predicted junction; you placed the FRET pair based on the predicted distance; you removed "redundant" residues at the fusion point based on the predicted contacts. All of these decisions were made on geometry that the network invented.
How to recognize the failure
Junction-region PAE. PAE across donor-domain boundaries is the single best diagnostic. Confidence within each donor is high; PAE between them is often >15 Å, betraying the artifact.
Sequence novelty at junction. If the junction sequence has no MSA hits, AF has no co-evolutionary information for it.
Multiple seeds disagree at junction. Re-run with different seeds; junctions in real fusions converge, junctions in artifacts diverge.
AlphaFold-Multimer as sanity check. Submitting the donor domains as separate chains often reveals that their preferred mutual orientation is not what the fused construct predicts.
What to trust, what not to
Trust | Don't trust |
|---|---|
Each donor domain's fold (especially if also confirmed by predicting each in isolation) | The junction geometry, linker conformation, or relative domain orientation |
Surface accessibility within each donor | FRET distances, BRET geometry, fluorophore orientation across the junction |
Likely PTM sites within each donor (AstraPTM) | Inter-domain allostery or signal propagation across the junction |
Remedy: For any chimera, use the Construct Design module to evaluate alternative linker lengths and compositions, then run a PAE Insight Engine pass on each candidate. The combination of AlphaFold2 integration with AstraUNFOLD-derived flexibility scores at the junction is the most reliable way to surface invented geometry before it becomes a cloning project.
Special case: circular permutants. Circular permutants are particularly treacherous. AlphaFold will fold the permuted sequence into something resembling the parent topology because the residue contacts are largely conserved. pLDDT often reads >85 throughout. But the new termini's effect on stability, the routing of the chain through the new break point, and the dynamics of the new loop region are essentially invented from scratch. Permutants designed solely from a confident AF prediction frequently fold partially, misfold, or are kinetically trapped during expression. Always validate permutant designs with FoldX or Rosetta stability calculations and with an explicit MSA-subsampled re-prediction.
When Confidence Scores Are Actually Reliable
It's important not to leave readers with the impression that pLDDT is broken. For the substantial majority of well-studied, single-domain, soluble proteins with deep MSAs, pLDDT is exactly what it claims to be: a strong predictor of local structural accuracy. The cases where it succeeds far outnumber the cases where it fails. The point is that the failure cases are concentrated in exactly the situations where structural biologists most often turn to AlphaFold: novel sequences, regulatory motifs, multi-state proteins, membrane targets, and engineered constructs.
A useful mental model: pLDDT is calibrated to the median protein in the PDB. The further your target is from that median—rarer sequence, conditional folding, multiple states, non-soluble compartment, non-natural construct—the more carefully you must interpret the score.
The contexts where pLDDT alone is genuinely sufficient:
Well-studied enzyme families with > 100 PDB structures in the same superfamily.
Single-domain soluble proteins from model organisms with deep MSAs (Neff/L > 5).
Antibody Fv regions (with the caveat that CDR3 loops remain difficult).
Tightly conserved structural folds (Ig sandwich, TIM barrel, Rossmann) in mainstream organisms.
The contexts where pLDDT must be paired with other evidence:
Anything outside the above categories.
Anything involving conditional folding, multi-state behavior, or membrane embedding.
Anything engineered, designed, fused, permuted, or otherwise non-natural.
Anything from an organism or family poorly represented in UniRef.
The Per-Residue pLDDT Interpretation Matrix
The standard pLDDT bands are well known. What's less appreciated is that the same pLDDT means very different things depending on context. The matrix below reflects this.
pLDDT band | If MSA is deep (Neff/L > 3) | If MSA is shallow (Neff/L < 1) | If region is a known MoRF | If region is in a TM helix | If region is in an engineered junction |
|---|---|---|---|---|---|
> 90 | Trust local geometry | Trust topology only, not contacts | Likely shows bound conformation; don't assume in solution | Helix exists; topology may not | Donor-domain interior is reliable; junction is not |
70–90 | Backbone reliable, rotamers vary | Treat as approximate fold | Strong MoRF suspicion | Helix likely real; orientation uncertain | Suspicious—often invented geometry |
50–70 | Approximate fold | Possibly fold-family default; verify | Often the true disordered baseline | Possibly re-entrant or amphipathic | Likely invented; do not use |
< 50 | Genuine disorder or genuine error | Lack of signal, not necessarily disorder | Expected for an apo MoRF | Often inter-helix loop—real disorder | Linker—often correct as "no structure" |
Decision Rules for Common Downstream Tasks
The reason these failure modes matter is that almost every downstream use of an AlphaFold model depends on the region-specific reliability of pLDDT. The following decision rules translate the matrix above into action.
For Construct Design
Boundaries should never be set inside a high-pLDDT region without checking whether that region is a MoRF (Case 2), a permuted junction (Case 6), or a loop pack artifact (Case 5).
Truncations should respect domain blocks defined by PAE, not by pLDDT alone.
Disordered tails (pLDDT < 50) are usually safe to remove; conditional-fold regions (pLDDT 70–88 with disorder-predictor disagreement) are not.
For Mutation Design
Single-point stability mutations in regions with pLDDT > 85 and local PAE < 5 Å are reasonable.
Mutations in MoRFs require knowledge of the bound partner; the predicted apo geometry is not a valid design target.
Active-site mutations in shallow-MSA proteins (Case 1) need experimental structure validation before being scaled into a library.
Domain-interface mutations require inter-domain PAE check, not just pLDDT at the residue of interest.
For Docking and Virtual Screening
Pocket geometry from AF2 is usable when pLDDT > 85, PAE < 5 Å within the pocket, and the protein is not a known multi-state target (Case 3).
For multi-state targets, dock against multiple AF predictions generated by MSA subsampling.
For membrane targets, validate topology before defining the docking site.
For Crystallization and Cryo-EM Construct Optimization
Use the PAE matrix, not pLDDT, to find the rigid blocks to crystallize.
Strip MoRFs and conditionally-folded tails before crystallization; they often prevent lattice formation.
For cryo-EM, low pLDDT regions are usually invisible in EM maps anyway, but they may interfere with particle picking if they cause aggregation.
For Mass Spec / HDX / Cross-linking Validation
Use confidence scores to predict where exchange should be slow (buried, high pLDDT, low PAE) vs fast (exposed, low pLDDT, high PAE).
Disagreement between predicted and observed HDX is a high-value signal that the AF model is wrong in that region.
Pitfall, Diagnostic, Remedy
Pitfall | Diagnostic | Remedy |
|---|---|---|
Shallow-MSA fold inheritance | Neff/L < 1; multi-seed disagreement; ESM-Fold disagreement | Subsampled MSA ensembles; orthogonal predictor comparison; SAXS |
Disorder-to-order trap | Composition-based disorder predictor disagrees with pLDDT; PAE asymmetry | AstraUNFOLD; AF-Multimer with known partner; CD/NMR |
Multi-state collapse | Multiple ranks differ; known fold-switcher family; SEC heterogeneity | AF-cluster; native MS; HDX-MS; NMR |
Membrane topology error | DeepTMHMM disagreement; polar residues on lipid face; inter-helix PAE high | Topology predictor consensus; PAE Insight Engine; compare against family |
Loop pack artifact | Long loop + charged composition; PAE to helices > 10 Å | HDX-MS; crystal B-factors of homologs; ensemble prediction |
Chimera junction artifact | Junction PAE > 15 Å; sequence novelty; multi-seed divergence | Construct Design module; AF-Multimer on donor pairs; linker scan |
A Cross-Validation Checklist Before Designing Experiments
Before any AlphaFold model becomes the basis for a wet-lab decision, run this checklist:
Per-residue pLDDT profile. Plot pLDDT vs residue. Identify all regions with pLDDT < 70 and treat them as suspect. Identify any sharp transitions—these are often domain boundaries or order-disorder transitions.
PAE matrix inspection. Open the PAE heatmap. Identify the diagonal blocks (rigid units). Check off-diagonal blocks for inter-domain confidence. Note any block at high pLDDT but high off-diagonal PAE—this is the "domains correct, arrangement wrong" signature.
MSA depth. Check the Neff/L of the input MSA. Below 1 is a yellow flag, below 0.3 is a red flag. If your protein is from an underrepresented clade or is de novo designed, expect prior-dominated predictions.
Orthogonal disorder check. Run IUPred3, PONDR-VSL2, or AstraUNFOLD on the same sequence. Wherever they disagree with pLDDT, flag the region as conditionally folded or potentially MoRF.
Multi-seed reproducibility. Re-run with 3–5 different seeds. Topological agreement at high pLDDT is a strong correctness signal. Topological disagreement at high pLDDT is the bistability or invention signature.
Family check. If the protein has paralogs with experimental structures, superpose them. RMSD > 3 Å in regions of high pLDDT is meaningful.
Topology predictor consensus (for membrane proteins). Check that AF's TM helix count and orientation match DeepTMHMM, Phobius, and TOPCONS.
If a region survives all seven checks, it's reasonable to design experiments on it. If it fails any of them, the failure should be addressed before, not after, you commit to a construct or library.
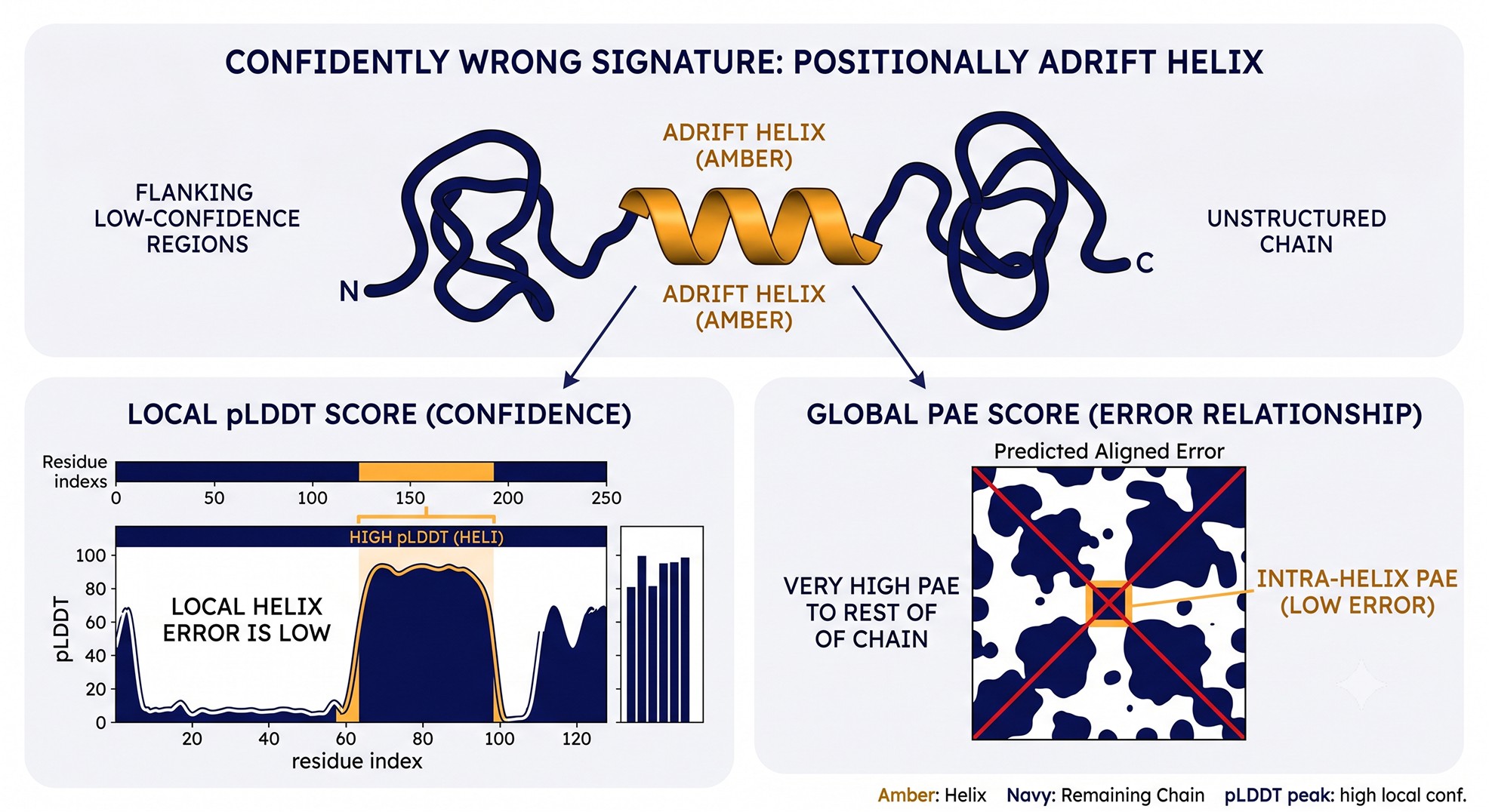
A Worked Example: The "Confidently Wrong" Helix
A representative scenario combining several of the cases above:
380-residue protein, single-domain prediction from AF2.
Average pLDDT = 81. Looks great.
Residues 240–262 are a deep-blue α-helix at mean pLDDT 87.
The researcher truncates at residue 240–262 as a stable "core" for crystallization. The construct expresses solubly but never crystallizes; CD shows ~30% helical content (expected ~70%); SEC shows polydispersity.
What the PAE matrix showed (and the researcher didn't check):
Residues 240–262 had intra-helix PAE of 4 Å (locally consistent).
Residues 240–262 had inter-region PAE to residues 1–239 of 22 Å (positionally adrift).
The N-terminal 80 residues had pLDDT 55–65 (overlooked because the helix was confident).
What was really going on: the helix is a MoRF that folds on partner binding (Case 2). The N-terminal 80 residues are a flexible regulatory segment that normally caps it (partially Case 5, partially Case 2). The confident helix in isolation is biologically meaningless. The construct boundary should have been residue 80–380, or the construct should have been co-expressed with its binding partner.
Cost of the mistake: ~4 months and 12 constructs. Cost of running PAE + AstraUNFOLD + AstraBIND beforehand: ~20 minutes.
The general lesson: any time pLDDT is high in a short segment whose flanking regions are low, the segment is structurally suspect even if locally consistent. Short, isolated, high-pLDDT helices in otherwise disordered regions are the canonical MoRF signature.
A Second Worked Example: The Membrane Transporter With Inverted Topology
A 12-TM transporter from a non-model bacterium was predicted by AF2 with average pLDDT of 89. Every TM helix scored above 90. The lab designed a C-terminal His-tag construct based on the implied topology (C-out, expecting external IMAC accessibility after detergent extraction). The construct expressed but did not bind IMAC under non-denaturing conditions, behaved aberrantly on BN-PAGE, and crystallization screens were uniformly precipitate.
The errors, visible only after running orthogonal topology analysis:
DeepTMHMM predicted 10 TM helices, not 12. AF had folded two amphipathic helices as full TM passes.
The positive-inside rule (von Heijne) placed two of the "TM" helices on the cytoplasmic side, not in the membrane.
The annotated N-glycosylation site (from the closely related eukaryotic homolog used as a reference) sat on the wrong face of the predicted structure.
Inter-helix PAE in the AF model was high (14–22 Å between several helix pairs), indicating that the bundle's geometry was uncertain even though each helix was locally confident.
The corrected construct used a different tagging scheme based on the topology-predictor consensus and crystallized within two rounds. The full diagnosis took an afternoon. The failed construct cost six months.
The Bottom Line
Failure mode | What pLDDT says | What's actually true | Best diagnostic |
|---|---|---|---|
Shallow-MSA fold inheritance | 80–90 in core | Topology may be a memorized prior | Multi-seed + ESM-Fold + SAXS |
Disorder-to-order transition | 70–88 on a helix | Helix exists only in complex | Composition + AF-Multimer with partner |
Multi-state collapse | 90+ on one state | A second state is equally real | AF-cluster + native MS + HDX |
Membrane topology error | 90+ per TM helix | Topology / bundle arrangement wrong | Topology predictor consensus + PAE |
Loop pack artifact | 70–88 on loop | Loop is dynamic in solution | HDX-MS + crystal B-factors |
Engineered junction | 80+ in donor domains | Junction geometry invented | Junction PAE + multi-seed + linker scan |
The single rule that prevents all six failures: never let pLDDT alone drive a decision that costs more than a day of work. Cross-reference with PAE, with an orthogonal disorder predictor, with topology software for membrane proteins, with multi-seed and subsampled-MSA ensembles for novel folds, and with experimental hydrodynamics or HDX-MS for anything that hits the bench. Confidence is not correctness; it is self-consistency. The two coincide most of the time, but the cases where they diverge are exactly the cases that cost months.
Frequently Asked Questions From Reviewers and Collaborators
"My average pLDDT is 87. Doesn't that mean the structure is reliable?"
Average pLDDT is the single least useful statistic AF2 emits. A 500-residue protein with 350 residues at 95 and 150 at 65 averages to 86 and hides a 30% unreliable surface area. Always read the per-residue profile.
"Why does my model look so confident when I know my protein is disordered?"
Almost certainly Case 1 (fold inheritance) or Case 2 (conditional folding). Cross-check with IUPred3 and AstraUNFOLD. If they disagree with AF, the disorder predictors are usually right for IDRs and MoRFs.
"AlphaFold says high pLDDT but the homologous crystal structure differs by 4 Å in this loop. Who's right?"
For loops, often the crystal structure represents one of several conformations the loop samples. Both can be "right" in different states. HDX-MS or NMR resolves the question.
"Should I use ipTM or pTM for a single-chain protein?"
Neither—pTM is the overall topology accuracy metric for single chains; pLDDT and PAE are the per-residue confidences you should rely on. ipTM is meaningful only for complexes.
"Why does subsampling the MSA give me a different structure?"
That's exactly what subsampling is designed to do—surface alternative conformations the network can produce. If subsampling gives stable alternatives, the protein may be genuinely multi-state (Case 3). If it gives noise, the prediction is fragile.
"My pLDDT increased after I added the binding partner in AF-Multimer. Is that real?"
Probably yes—and it's diagnostic. A region whose confidence increases on partner addition was likely a MoRF (Case 2). Use the holo prediction for the bound state, but be aware the apo state is conformationally different in solution.
How Orbion Surfaces These Failures Before They Cost You
Most pLDDT misinterpretation traps share a common root: the score is informative in isolation, but only safe in combination. Orbion's structural confidence workflow is built around that combination. The PAE Insight Engine reads the full PAE matrix from AlphaFold2 / Multimer integration and surfaces the three signatures most predictive of the failure modes above: intra-region/inter-region PAE asymmetry, inter-helix PAE in TM bundles, and junction PAE in engineered constructs. AstraUNFOLD overlays composition-based disorder, conditional-fold (MoRF/SLiM) propensity, and topology calls so that a confident helix flagged as a likely MoRF is highlighted before you cut a construct around it. For chimeras and fusions, the Construct Design module reports per-residue confidence and PAE at every candidate linker boundary, so you can see whether the junction geometry is real or invented. The net effect: pLDDT becomes one input among several, never the single driver of a costly downstream decision.
References
Jumper J, et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596:583–589. Link
Tunyasuvunakool K, et al. (2021). Highly accurate protein structure prediction for the human proteome. Nature, 596:590–596. Link
Ruff KM & Pappu RV. (2021). AlphaFold and Implications for Intrinsically Disordered Proteins. Journal of Molecular Biology, 433(20):167208. Link
Buel GR & Walters KJ. (2022). Can AlphaFold2 predict the impact of missense mutations on structure? Nature Structural & Molecular Biology, 29:1–2. Link
Saldaño T, et al. (2022). Impact of protein conformational diversity on AlphaFold predictions. Bioinformatics, 38(10):2742–2748. Link
Akdel M, et al. (2022). A structural biology community assessment of AlphaFold2 applications. Nature Structural & Molecular Biology, 29:1056–1067. Link
Mariani V, et al. (2013). lDDT: a local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics, 29(21):2722–2728. PMC3799472
Book a 20-Minute Demo
Sign up free for unlimited Overview runs — summary, sequence-based analysis, homology search. For the full Characterization — PTMs, binding sites, stability variants, construct design — book a demo and we'll run your target live.
